453/2004 Sb.
NAŘÍZENÍ VLÁDY
ze dne 7. července 2004,
kterým se stanoví technické požadavky na diagnostické zdravotnické prostředky
in vitro
Změna: 246/2009 Sb.
Změna: 67/2011 Sb.
Změna: 223/2012 Sb.
Vláda nařizuje podle § 22 zákona č. 22/1997 Sb., o technických požadavcích na
výrobky a o změně a doplnění některých zákonů, ve znění zákona č. 71/2000 Sb. a zákona
č. 205/2002 Sb., (dále jen "zákon") k provedení § 11, 12 a 13 zákona a k provedení
zákona č. 123/2000 Sb., o zdravotnických prostředcích a o změně některých souvisejících
zákonů, ve znění zákona č. 130/2003 Sb. a zákona č. 274/2003 Sb., (dále jen "zákon
o zdravotnických prostředcích"):
§ 1
Toto nařízení upravuje v návaznosti na přímo použitelný předpis Evropské
unie1)
a) technické požadavky na diagnostické zdravotnické prostředky in vitro2)
(dále jen "in vitro diagnostika") a
b) společné technické specifikace pro in vitro diagnostika.
§ 2
(1) In vitro diagnostika jsou stanovenými výrobky podle § 12 odst. 1 zákona,
u kterých se posuzuje shoda jejich vlastností podle tohoto nařízení.
(2) Toto nařízení se použije i pro
a) in vitro diagnostika vyrobená a určená k použití pro účely rozborů v
klinických laboratořích, aniž jsou předmětem obchodování,
b) mechanická laboratorní zařízení zvláště navržená pro diagnostická vyšetření
in vitro,
c) in vitro diagnostika vyrobená z tkání, buněk nebo látek lidského původu,
d) příslušenství in vitro diagnostik, se kterými se zachází jako se samostatnými
in vitro diagnostiky,
e) in vitro diagnostika, kterými jsou kalibrátory a kontrolní materiály
potřebné k určení nebo ověření funkční způsobilosti jejich uživatelem3).
(3) Toto nařízení se nevztahuje na
a) nástroje, přístroje, zařízení nebo jiné předměty včetně programového
vybavení určené k použití pro výzkumné účely bez jakéhokoliv lékařského cíle,
b) mezinárodně osvědčené referenční materiály a materiály používané pro
programy externího posuzování systémů jakosti,
c) činidla, která jsou vyrobena v laboratořích zdravotnického zařízení
pro účely použití v prostředí těchto laboratoří a nejsou předmětem obchodování,
d) zdravotnické prostředky4) pro invazivní odběr vzorků z lidského těla,
e) in vitro diagnostika, která byla vyrobena ve zdravotnickém zařízení
a používají se pouze v tomto zdravotnickém zařízení, aniž byla předána jinému uživateli,
f) jiné zdravotnické prostředky vyrobené za použití látek lidského původu
[s výjimkou in vitro diagnostik uvedených v odstavci 2 písm. c)].
§ 3
Výklad pojmů
Pro účely tohoto nařízení se dále rozumí
a) in vitro diagnostiky pro sebetestování in vitro diagnostika, která jsou
určena výrobcem k použití laikem v domácím prostředí,
b) funkční způsobilostí souhrn vlastností in vitro diagnostik stanovený
výrobcem pro jím určený účel použití těchto in vitro diagnostik,
c) in vitro diagnostikem pro ověření funkční způsobilosti in vitro diagnostikum
určené výrobcem pro ověření funkční způsobilosti prováděné v klinických laboratořích
nebo v jiných odpovídajících prostředích; tato činnost se neprovádí v objektech výrobce,
d) zplnomocněným zástupcem osoba usazená v členském státě Evropské unie
nebo Evropského sdružení volného obchodu, který je současně smluvní stranou Evropského
hospodářského prostoru, (dále jen „členský stát“), která je výrobcem písemně pověřena
k jednání za něj s úřady a orgány v členských státech, se zřetelem na požadavky vyplývající
pro výrobce z tohoto nařízení,
e) uvedením na trh okamžik, kdy in vitro diagnostika, která nejsou určena
pro ověření funkční způsobilosti, přecházejí poprvé úplatně nebo bezúplatně do distribuce
jako zboží určené k prodeji v členských státech, bez ohledu na to, zda jsou nová
nebo plně obnovená,
f) uvedením do provozu okamžik, kdy in vitro diagnostika připravená poprvé
pro stanovený účel použití jsou poskytnuta konečnému uživateli,
g) kalibračními a kontrolními materiály látky, materiály nebo předměty
určené jejich výrobcem ke stanovení rozsahu měření nebo k ověření funkční způsobilosti
in vitro diagnostik v souladu s jejich určeným účelem použití,
h) příslušenstvím předmět, který není in vitro diagnostikem, ale je určen
výrobcem specificky k použití společně s in vitro diagnostikem tak, aby umožnil jeho
použití v souladu s účelem, který mu výrobce určil; za příslušenství in vitro diagnostik
se nepovažují zdravotnické prostředky pro invazivní odběr vzorků z lidského těla,
i) novým in vitro diagnostikem takové, které
1. během předcházejících
3 let nebylo pro příslušný analyt nebo jiný parametr nepřetržitě k dispozici na trhu
v členských státech,
2. využívá postup analytické technologie, která během předcházejících
3 let nebyla nepřetržitě využívána v souvislosti s daným analytem nebo jiným parametrem
na trhu v členských státech.
§ 4
Obecné zásady
(1) In vitro diagnostika musí vyhovovat požadavkům uvedeným v příloze č. 1
k tomuto nařízení, které se na ně vztahují, (dále jen "základní požadavky") s přihlédnutím
k určenému účelu použití. Základní požadavky se považují rovněž za splněné, jestliže
in vitro diagnostika odpovídají příslušným harmonizovaným normám5); za harmonizované
normy se podle tohoto nařízení považují i společné technické specifikace pro in vitro
diagnostika6).
(2) Při odebírání vzorků z lidského organismu (dále jen "vzorky") a při odběru
a použití látek pocházejících z lidského těla se postupuje v souladu s mezinárodní
smlouvou7) a etickými zásadami8).
(3) Jestliže in vitro diagnostika uvedená v § 5 odst. 1 a 2 jsou řádně
instalovaná, udržovaná a používaná v souladu s určeným účelem použití, a přesto mohou
ohrozit zdraví, popřípadě život uživatelů nebo majetek, postupuje se podle zákona,
zákona o zdravotnických prostředcích a podle zvláštního právního předpisu9). Za důvod
k takovému postupu se považuje zejména
a) nedodržení základních požadavků uvedených podle odstavce 1,
b) nesprávné použití harmonizovaných norem, pokud se prohlašuje, že byly
použity, nebo
c) nedostatek v samotných harmonizovaných normách.
(4) Informace poskytované uživateli podle bodu 8 části B přílohy č. 1 k tomuto
nařízení musí být v českém jazyce, pokud nejde o symboly uvedené v příloze č. 1 k
tomuto nařízení nebo v příslušné harmonizované normě10).
§ 5
Uvedení in vitro diagnostik na trh a do provozu
(1) In vitro diagnostika mohou být uvedena na trh11), jestliže
a) byla u nich posouzena shoda v souladu s § 8,
b) splňují základní požadavky a další ustanovení tohoto nařízení, která
se na ně vztahují,
c) bylo pro ně vydáno prohlášení o shodě podle tohoto nařízení a
d) byla opatřena označením CE12).
(2) Do provozu mohou být in vitro diagnostika uvedena pouze tehdy, jestliže
splňují podmínky uvedené v odstavci 1 a byla dodána a instalována odpovídajícím způsobem
v souladu s určeným účelem použití.
(3) In vitro diagnostika po uvedení na trh musí být sledována z hlediska jejich
bezpečnosti a kvality v souladu s § 12.
(4) Ustanovení odstavců 1 až 3 se vztahují rovněž na in vitro diagnostika poskytnutá
pro ověření jejich funkční způsobilosti laboratořím nebo jiným osobám, které splňují
požadavky stanovené v § 8 odst. 4 a v příloze č. 8 k tomuto nařízení.
§ 6
Vystavování in vitro diagnostik
Předvádět na výstavách, veletrzích i jinak in vitro diagnostika, která neodpovídají
požadavkům tohoto nařízení, lze jen za předpokladu, že jsou viditelně označena tak,
aby bylo patrné, že nemohou být uváděna na trh a do provozu, dokud nebudou uvedena
do souladu s požadavky tohoto nařízení; pro předvádění in vitro diagnostik nesmí
být použity vzorky odebrané od účastníků tohoto předvádění.
§ 7
Společné technické specifikace
(1) Společné technické specifikace stanoví
a) kritéria pro
1. ověření a přehodnocení funkční způsobilosti in vitro
diagnostik a
2. uvolňování výrobních šarží,
b) referenční
1. metodiky a
2. materiály,
a to u in vitro diagnostik uvedených v Seznamu A přílohy č. 2 k tomuto
nařízení, a kde je to nutné, u in vitro diagnostik uvedených v Seznamu B přílohy
č. 2 k tomuto nařízení.
(2) Společné technické specifikace lze v řádně odůvodněných případech při návrhu
a výrobě in vitro diagnostik nahradit jiným řešením, kterým se dosáhne přinejmenším
stejné úrovně bezpečnosti a kvality in vitro diagnostik.
(3) Společné technické specifikace jsou stanoveny v přímo použitelném právním
předpisu Evropské unie12a).
(4) Jestliže společnými technickými specifikacemi nelze zcela ověřit splnění
základních požadavků, postupuje se podle zákona13).
§ 8
Postupy posuzování shody
(1) Před uvedením in vitro diagnostik na trh, s výjimkou těch, které jsou uvedeny
v příloze č. 2 k tomuto nařízení, a těch, které jsou určeny pro ověřování funkční
způsobilosti, postupuje výrobce při opatřování označením CE v souladu s § 9 a podle
přílohy č. 3 k tomuto nařízení a vyhotoví písemné ES prohlášení o shodě. Výrobce
u in vitro diagnostik pro sebetestování před vyhotovením písemného ES prohlášení
o shodě plní další požadavky stanovené v bodě 6 přílohy č. 3 k tomuto nařízení nebo
postupuje podle odstavce 2 nebo 3.
(2) U in vitro diagnostik uvedených v Seznamu A přílohy č. 2 k tomuto nařízení,
s výjimkou in vitro diagnostik určených pro ověření funkční způsobilosti, výrobce
postupuje za účelem opatřování označením CE v souladu s § 9 podle
a) přílohy č. 4 k tomuto nařízení, nebo
b) přílohy č. 5 k tomuto nařízení společně s postupem podle přílohy č.
7 k tomuto nařízení.
(3) U in vitro diagnostik uvedených v Seznamu B přílohy č. 2 k tomuto nařízení,
s výjimkou in vitro diagnostik pro ověření funkční způsobilosti, výrobce postupuje
za účelem opatřování označením CE v souladu s § 9 podle
a) přílohy č. 4 k tomuto nařízení, nebo
b) přílohy č. 5 k tomuto nařízení společně s postupem podle přílohy č.
6 nebo podle přílohy č. 7 k tomuto nařízení.
(4) V případech in vitro diagnostik pro ověření funkční způsobilosti výrobce
postupuje podle přílohy č. 8 k tomuto nařízení a před jejich zpřístupněním vypracuje
písemné prohlášení stanovené touto přílohou. Tímto ustanovením nejsou dotčeny předpisy
týkající se etických hledisek8) při provádění studií ověřujících funkční způsobilost
za použití tkání nebo látek lidského původu.
(5) Při posuzování shody in vitro diagnostik výrobce, popřípadě notifikovaná
osoba14) zohledňuje výsledky hodnocení a ověřovacích postupů, pokud byly ve vhodných
případech provedeny v souladu s tímto nařízením v jednotlivých mezistupních výrobního
procesu.
(6) Výrobce po dobu 5 let po vyrobení posledního in vitro diagnostika uchovává
prohlášení o shodě, technickou dokumentaci uvedenou v přílohách č. 3 až 8 k tomuto
nařízení, zprávy, certifikáty, popřípadě jiná rozhodnutí vyhotovená notifikovanými
osobami a zpřístupňuje je příslušným orgánům státní správy pro kontrolní účely.
(7) Výrobce může vydat pokyn svému zplnomocněnému zástupci, aby zahájil postupy
uvedené v přílohách č. 3, 5, 6 a 8 k tomuto nařízení.
(8) Jestliže dokumentaci podle odstavce 6 neposkytl výrobce, poskytne ji na
žádost zplnomocněný zástupce, byl-li výrobcem ustanoven.
(9) Zahrnuje-li postup posuzování shody účast notifikované osoby, pak výrobce,
popřípadě jeho zplnomocněný zástupce požádá o tuto účast notifikovanou osobu s odpovídajícím
rozsahem autorizace. Notifikovaná osoba může požadovat nezbytně nutné informace nebo
údaje pro schválení a dodržování zvoleného postupu posuzování shody.
(10) Certifikáty notifikované osoby vydané v souladu s přílohami č. 3, 4 a
5 k tomuto nařízení jsou platné po dobu nejdéle 5 let, jejich platnost může být prodloužena
o dalších 5 let, a to na základě žádosti výrobce.
(11) Záznamy a korespondence týkající se postupů podle odstavců 1 až 4 se pořizují
v českém jazyce, popřípadě v jiném jazyce, na kterém se dohodne notifikovaná osoba
s výrobcem, popřípadě se zplnomocněným zástupcem.
(12) Postup v případech, ve kterých se výjimečně u in vitro diagnostik nemusí
posoudit shoda, stanoví zákon o zdravotnických prostředcích15).
(13) Ustanovení odstavců 1 až 11 se přiměřeně vztahují na fyzické a právnické
osoby, které in vitro diagnostika vyrábějí a samy je uvádějí do provozu tak, že je
v rámci své profesní činnosti používají.
§ 9
Označování in vitro diagnostik
(1) In vitro diagnostika, jiná než pro ověřování funkční způsobilosti, která
splňují základní požadavky, musí být před uvedením na trh opatřena označením CE.
(2) Označení in vitro diagnostika podle odstavce 1 musí být umístěno viditelně,
čitelně a nesmazatelně, pokud je to proveditelné a vhodné, a dále v návodu k použití.
K označení CE musí být připojeno identifikační číslo notifikované osoby podílející
se na postupech stanovených v přílohách č. 3, 4, 6 a 7 k tomuto nařízení.
(3) Na in vitro diagnostika nesmějí být umístěny značky a popisy, které by
omylem mohly být považovány za znaky významem nebo graficky se podobající označení
CE. Jiný znak lze umístit na in vitro diagnostika, jejich obal nebo v návodech doprovázejících
tyto in vitro diagnostika za předpokladu, že tím není snížena viditelnost ani čitelnost
označení CE.
(4) Jednotlivé části označení CE u diagnostických zdravotnických prostředků
in vitro musí mít zásadně stejnou výšku, která nesmí být menší než 5 mm. Tento minimální
rozměr může být pro in vitro diagnostika malých rozměrů upraven.
(5) Pokud in vitro diagnostika podléhají z dalších hledisek zvláštním právním
předpisům, které stanoví povinnost je opatřit označením CE, vyjadřují v takovém případě
tato označení, že in vitro diagnostika jsou v souladu také s požadavky, které se
na ně vztahují podle těchto zvláštních právních předpisů. Jestliže však jeden nebo
několik právních předpisů po přechodnou dobu připouští, aby výrobce zvolil, kterými
ustanoveními se bude řídit, pak označení CE vyjadřuje shodu pouze s těmi právními
předpisy nebo jejich ustanoveními, které výrobce použil. V tomto případě musí být
v dokumentaci, upozorněních nebo návodech, požadovaných příslušnými právními předpisy,
a přiložených k příslušným výrobkům, uveden seznam použitých právních předpisů a
dále směrnic, jak byly uveřejněny v Úředním věstníku Evropské unie, jejichž požadavky
byly těmito právními předpisy převzaty.
§ 10
Nesprávné použití označení CE
(1) V případě zjištění, že in vitro diagnostikum bylo nesprávně opatřeno označením
CE, výrobce nebo jeho zplnomocněný zástupce odstraní tento protiprávní stav podle
pokynů České obchodní inspekce16).
(2) Jestliže nebyl odstraněn protiprávní stav podle odstavce 1, postupuje se
podle zvláštních právních předpisů17).
(3) Ustanovení odstavců 1 a 2 se vztahují i na případy, kdy výrobky byly opatřeny
označením CE v souladu s postupy podle tohoto nařízení, avšak tyto výrobky nespadají
do působnosti tohoto nařízení.
§ 11
Oznamovací povinnosti
(1) Výrobce, který v souladu s postupy podle § 5 uvádí in vitro diagnostika
na trh nebo do provozu, nebo zplnomocněný zástupce oznamuje v elektronické podobě
Ministerstvu zdravotnictví (dále jen „ministerstvo“)
a) zahájení činnosti v souladu
s přílohou č. 11 k tomuto nařízení,
b) ukončení činnosti v souladu s přílohou č. 11
k tomuto nařízení,
c) uvedení in vitro diagnostika na trh v souladu s přílohou č.
13 k tomuto nařízení, a to ještě před uvedením in vitro diagnostika na trh,
d) ukončení
uvádění in vitro diagnostika na trh v souladu s přílohou č. 13 k tomuto nařízení
a
e) změnu některého z údajů oznámených podle písmen a) a c).
Údaje podle přílohy č.
13 k tomuto nařízení se oznamují až poté, co oznamovatel obdrží od ministerstva evidenční
číslo na základě splnění oznamovací povinnosti v souladu s přílohou č. 11 k tomuto
nařízení.(2) Předmětem oznámení podle odstavce 1 je rovněž nové in vitro diagnostikum,
které bylo opatřeno označením CE.
(3) Osoba, uvádějící in vitro diagnostika na trh nebo do provozu na území České
republiky, je povinna na žádost ministerstva poskytnout informace umožňující identifikaci
in vitro diagnostika podle bodu 2. přílohy č. 13 k tomuto nařízení a podklady, které
sloužily k posouzení shody.
(4) Jestliže výrobce, který zamýšlí uvést pod svým jménem na trh in vitro diagnostika
podle odstavce 1 nebo 2, nemá sídlo v členském státě, pověří pro uvádění těchto in
vitro diagnostik na trh zplnomocněného zástupce.
(5) Do zprovoznění Evropské databanky zdravotnických prostředků18) oznamuje
ministerstvu informace podle odstavců 1 až 3 i výrobce se sídlem v jiném členském
státě než v České republice, pokud uvádí in vitro diagnostika na trh v České republice.
(6) Distributor, dovozce a osoba provádějící servis in vitro diagnostik
oznamují v elektronické podobě ministerstvu
a) zahájení činnosti v souladu s přílohou č. 12 k tomuto nařízení,
b) ukončení činnosti v souladu s přílohou č. 12 k tomuto nařízení a
c) změnu některého z údajů oznámených podle písmene a).
§ 12
Postup při předcházení nežádoucím příhodám a při jejich vzniku
(1) Při předcházení nežádoucím příhodám a při jejich vzniku se postupuje v
souladu s ustanoveními § 4 odst. 3 a zákona o zdravotnických prostředcích19).
(2) Pokud jde o nežádoucí příhodu u nového in vitro diagnostika, ke kterému
bylo připojeno označení CE, výrobce tuto skutečnost uvede v hlášení o nežádoucí příhodě20).
(3) Státní ústav pro kontrolu léčiv (dále jen "Ústav") může požadovat v odůvodněných
případech do 2 let po ohlášení nežádoucí příhody informace o zkušenostech s novým
in vitro diagnostikem po jeho uvedení na trh.
§ 13
Soubor údajů
(1) Údaje
a) o in vitro diagnostikách a osobách uvedených v § 11,
b) o vydaných,
změněných, pozastavených a zrušených certifikátech, jakož i o zamítnutí žádostí o
vydání certifikátu podle postupů stanovených v přílohách č. 3 až 7 k tomuto nařízení
a
c) získané v souladu s postupem upravujícím oznamování a evidenci nežádoucích příhod21)
se
zpracovávají v souladu s tímto nařízením v informačním systému podle zákona o zdravotnických
prostředcích22) do doby, kdy osoba nakládající s in vitro diagnostiky oznámí ministerstvu
ukončení činnosti, a po dobu dalších 20 let. Údaje z informačního systému jsou přístupné
ministerstvu, Úřadu pro technickou normalizaci, metrologii a státní zkušebnictví
(dále jen „Úřad“), Ústavu, Státnímu úřadu pro jadernou bezpečnost u zdrojů ionizujícího
záření, Ústavu zdravotnických informací a statistiky České republiky a České obchodní
inspekci. Údaje z informačního systému o nežádoucích příhodách21) in vitro diagnostik
jsou přístupné pouze ministerstvu, Ústavu a Ústavu zdravotnických informací a statistiky
České republiky.(2) Údaje uvedené v odstavci 1 písm. a) se poskytují v elektronické podobě
podle příloh č. 11 až 13 k tomuto nařízení.
§ 14
Ochranná opatření a úprava seznamu in vitro diagnostik podle přílohy č. 2
(1) V případě zjištění, že in vitro diagnostikům by měla být pro zajištění
bezpečnosti a ochrany zdraví, popřípadě veřejného zdraví23), zakázána nebo omezena
jejich dostupnost, anebo by in vitro diagnostika měla podléhat zvláštním požadavkům,
přijímají ministerstvo, Ústav, Česká obchodní inspekce, popřípadě další příslušné
úřady na přechodnou dobu odpovídající opatření podle zvláštních právních předpisů24);
těmito opatřeními jsou zejména omezení či zákaz uvádění in vitro diagnostik na trh,
stažení z trhu, nebo omezení a zákaz používání při poskytování zdravotní péče. Tato
opatření musí obsahovat přesné důvody, na nichž se zakládají; opatření se neprodleně
oznamují tomu, koho se bezprostředně dotýkají, a to spolu s informací o opravných
prostředcích a lhůtách, v nichž lze opravné prostředky podat.
(2) V době přípravy opatření podle odstavce 1 může výrobce nebo zplnomocněný
zástupce sdělit k němu své stanovisko, pokud nejde o nebezpečí z prodlení s ohledem
na ochranu zdraví24).
(3) Pokud ministerstvo dospěje k názoru, že by
a) seznam in vitro diagnostik uvedených v příloze č. 2 k tomuto nařízení
měl být opraven nebo rozšířen, nebo
b) shoda in vitro diagnostik nebo jejich kategorie by měly být stanoveny
odchylně, a to použitím jednoho nebo více postupů vybraných podle § 8,
předloží Evropské komisi řádně zdůvodněnou žádost o rozhodnutí.
(4) Před předložením žádosti podle odstavce 3 je třeba zohlednit
a) jakékoliv podstatné informace dostupné z postupů při předcházení nežádoucím
příhodám nebo při jejich vzniku a z programů externího posuzování jakosti,
b) zda
1. se lze přesně spolehnout na výsledek získaný daným in vitro
diagnostikem vzhledem k tomu, že tento výsledek má přímý vliv na následný lékařský
výkon,
2. výkon učiněný na základě nesprávného výsledku získaného použitím in vitro
diagnostika by se mohl ukázat jako nebezpečný pro pacienta a další osoby, zejména
následkem nesprávně pozitivních nebo nesprávně negativních výsledků, a
3. účast
notifikované osoby by přispěla k zajištění shody in vitro diagnostika.
§ 15
Notifikované osoby
(1) Podmínky autorizace jsou stanoveny v příloze č. 9 k tomuto nařízení; o
právnických osobách, které vyhoví požadavkům českých technických norem, přebírají-li
plně požadavky stanovené evropskou normou nebo harmonizačním dokumentem, se předpokládá,
že vyhovují příslušným kritériím pro autorizaci.
(2) Notifikovaná osoba a výrobce nebo jeho zplnomocněný zástupce na základě
dohody stanoví termíny pro dokončení hodnotících a ověřovacích činností uvedených
v přílohách č. 3 až 7 k tomuto nařízení.
(3) Notifikovaná osoba informuje ostatní notifikované osoby a Úřad o pozastavení
nebo zrušení certifikátů, které vydala; na požádání informuje ostatní notifikované
osoby a Úřad o vydání certifikátu nebo zamítnutí žádosti o vydání certifikátů a poskytne
další podstatné informace.
(4) V případě, že notifikovaná osoba zjistí, že požadavky podle tohoto nařízení
nebyly výrobcem splněny nebo již nadále nejsou plněny, nebo v případech, kdy certifikát
neměl být vydán, notifikovaná osoba, s přihlédnutím k zásadám přiměřenosti, pozastaví
nebo zruší vydaný certifikát nebo omezí jeho použití, pokud výrobce nezajistí shodu
s těmito požadavky zavedením odpovídajících nápravných opatření. V případě pozastavení,
omezení nebo zrušení certifikátu nebo v případech, kdy může být nutný zásah Úřadu,
notifikovaná osoba o této skutečnosti informuje Úřad.
(5) Při informování příslušných úřadů jiných států a Evropské komise o certifikátech,
které notifikovaná osoba změnila nebo zrušila podle předchozích odstavců, se postupuje
podle zákona nebo zákona o zdravotnických prostředcích podle jejich působnosti.
(6) Notifikovaná osoba poskytne na vyžádání Úřadu podstatné informace a dokumenty,
včetně rozpočtových dokumentů, k ověření kritérií uvedených v příloze č. 9 k tomuto
nařízení.
§ 16
Přechodná ustanovení
(1) In vitro diagnostika, která splňují požadavky podle dosavadních právních
předpisů25), mohou být uváděna do provozu nejdéle do 7. prosince 2005.
(2) In vitro diagnostika registrovaná podle § 11 nařízení vlády č. 286/2001
Sb., kterým se stanoví technické požadavky na diagnostické zdravotnické prostředky
in vitro, se považují za in vitro diagnostika oznámená podle § 11.
(3) In vitro diagnostika, u nichž byla posouzena shoda podle § 8 nařízení vlády
č. 286/2001 Sb., kterým se stanoví technické požadavky na diagnostické zdravotnické
prostředky in vitro, lze uvádět na území České republiky na trh nejdéle do 7. prosince
2005, a to pro uvedení do provozu pouze v České republice.
§ 17
Zrušovací ustanovení
Nařízení vlády č. 286/2001 Sb., kterým se stanoví technické požadavky na diagnostické
zdravotnické prostředky in vitro, se zrušuje.
Předseda vlády:
PhDr. Špidla v. r.
Ministr zdravotnictví:
MUDr. Kubinyi, Ph.D. v. r.
Příl.1
ZÁKLADNÍ POŽADAVKY
A.
VŠEOBECNÉ POŽADAVKY
1. In vitro diagnostika musí být navržena a vyrobena tak, aby jejich použití
za stanovených podmínek a k určenému účelu neohrozila, přímo nebo nepřímo, klinický
stav, bezpečnost nebo zdraví uživatelů, popřípadě majetek. Jakákoliv rizika, která
mohou v souvislosti s jejich použitím vzniknout, musí být přijatelná ve srovnání
s přínosem, který pro pacienty představují a tyto vlastnosti musí odpovídat vysoké
úrovni ochrany zdraví a bezpečnosti.
2. Konečné řešení, které výrobce zvolí pro návrh in vitro diagnostik,
musí vycházet ze stavu vědy a techniky odpovídající době, kdy bylo toto in vitro
diagnostikum vyrobeno. Při výběru nejvhodnějšího řešení výrobce dodržuje následující
zásady v uvedeném pořadí:
2.1. vyloučit nebo co nejvíce snížit rizika (bezpečným návrhem a konstrukcí),
2.2. učinit odpovídající ochranná opatření vůči rizikům, která nelze
vyloučit,
2.3. informovat uživatele v případě přetrvávání rizik, která nebyla
odstraněna z důvodu nedokonalých ochranných opatření.
3. In vitro diagnostika musí být navržena a vyrobena takovým způsobem, aby
byla vhodná pro účely uvedené v zákoně o zdravotnických prostředcích v souladu se
specifikací výrobce a zároveň musí respektovat stav vědy a techniky odpovídající
době, kdy byla vyrobena. Musí dosáhnout funkční způsobilosti stanovené výrobcem,
a to zejména přichází-li v úvahu citlivost k příslušnému rozboru, citlivost pro diagnózu,
analytickou specificitu, diagnostickou vhodnost, přesnost, opakovatelnost, reprodukovatelnost,
včetně kontroly týkající se vzájemné interference, rušivých vlivů, a stanovení meze
detekce, deklarovaných výrobcem.
Hodnoty, které jsou stanovené pro kalibrátory nebo
pro kontrolní materiály, musí být ověřeny pomocí dostupných referenčních metod měření,
případně dostupných materiálů vyšší úrovně.
4. Charakteristické vlastnosti a funkční způsobilost uvedené v ustanovení
bodů 1 až 3 se nesmí změnit tak, že by in vitro diagnostikum během doby použitelnosti
nebo životnosti stanovené výrobcem, ohrozilo zdraví a bezpečnost uživatele, ani v
případě, že in vitro diagnostikum je vystaveno nevhodným podmínkám nebo zatížení,
ke kterému může dojít i za normálních podmínek jeho používání. Pokud není doba použitelnosti
nebo životnost in vitro diagnostika stanovena, platí totéž pro dostatečně předvídatelnou
dobu použitelnosti nebo životnosti in vitro diagnostika tohoto typu s ohledem na
jeho určený účel a předpokládané použití.
5. In vitro diagnostika musí být navržena, vyrobena a zabalena takovým způsobem,
aby jejich vlastnosti a funkční způsobilost při určeném účelu použití nebyly nepříznivě
ovlivněny podmínkami při skladování nebo dopravě (teplotou, vlhkostí vzduchu a podobně),
přičemž musí být zachovány pokyny nebo informace určené výrobcem.
B.
POŽADAVKY NA NÁVRH A VÝROBU
1.
Chemické a fyzikální vlastnosti.
1.1. In vitro diagnostika musí být navržena a vyrobena takovým způsobem,
aby byly zabezpečeny charakteristiky a funkční způsobilost podle části A "Všeobecné
požadavky". Zvláštní pozornost musí být věnována možnosti snížení analytické funkční
způsobilosti z důvodu vzájemné nekompatibility mezi použitými materiály a vzorky
(např. biologickými tkáněmi, buňkami a mikroorganizmy), které jsou určeny k použití
spolu s in vitro diagnostikem k jeho určenému účelu.
1.2. In vitro diagnostika musí být navržena, vyrobena a zabalena tak,
aby byla minimalizována rizika úniku tekutin z výrobku, úniku kontaminujících látek
z kontaminovaných výrobků a dalších znečišťujících látek během skladování, přepravy
a použití in vitro diagnostik, přičemž se musí dodržovat pokyny určené výrobcem.
2.
Infekce a mikrobiální kontaminace.
2.1. In vitro diagnostika a výrobní postupy s nimi související musí být
navrženy tak, aby se pokud možno odstranilo nebo snížilo riziko infekce pro uživatele.
Návrh musí umožnit snadnou manipulaci, a pokud možno minimalizovat kontaminaci a
únik kapalin během použití. V případě nádob na vzorky je nutné snížit riziko znečištění
vzorku. Výrobní postupy musí odpovídat těmto účelům.
2.2. Obsahují-li in vitro diagnostika biologické látky, musí být minimalizováno
riziko infekce výběrem vhodného dárce a odpovídajících látek a použitím příslušných
validovaných inaktivačních, konzervačních, zkušebních a řídicích postupů.
2.3. In vitro diagnostika označená buď jako "STERILNÍ" nebo jako in vitro
diagnostika ve zvláštním mikrobiologickém stavu, musí být navržena, vyrobena a zabalena
v odpovídajícím obalu podle vhodných postupů, které zajišťují, že výrobcem stanovené
podmínky skladování a dopravy zůstanou až do poškození nebo otevření ochranného obalu
v mikrobiologickém stavu, který odpovídá značení uvedenému na in vitro diagnostiku
při jeho uvedení na trh.
2.4. In vitro diagnostika označená buď jako "STERILNÍ" nebo jako in vitro
diagnostika ve zvláštním mikrobiologickém stavu, musí být zpracována odpovídající
validovanou metodou.
2.5. Obalové systémy in vitro diagnostik kromě těch, které jsou uvedeny
v bodu 2.3. musí uchovat in vitro diagnostikum bez zhoršení stupně čistoty uvedené
výrobcem, a mají-li být in vitro diagnostika před použitím sterilizována, musí systém
v rozumné míře minimalizovat možné riziko mikrobiální kontaminace.
Dále musí být
učiněna opatření, která co nejvíce omezí možnost mikrobiálního znečištění při výběru
a manipulaci se surovinami, při výrobě, skladování a distribuci v případě, že by
funkční způsobilost in vitro diagnostik mohla být ovlivněna takovým znečištěním.
2.6. In vitro diagnostika, která mají být sterilizována, musí být vyrobena
za odpovídajících řízených podmínek (například podmínek prostředí).
2.7. Obalové systémy pro nesterilní in vitro diagnostika musí uchovávat
in vitro diagnostikum bez zhoršování kvality na určené úrovni čistoty, a jsou-li
in vitro diagnostika určena ke sterilizaci před použitím, musí minimalizovat riziko
mikrobiálního znečištění. Obalový systém musí být vhodný s ohledem na metodu sterilizace
uvedenou výrobcem.
3.
Vlastnosti in vitro diagnostik ve vztahu k výrobě a prostředí.
3.1. Je-li in vitro diagnostikum určeno k použití v kombinaci s jinými
in vitro diagnostiky nebo vybavením, musí být celá kombinace včetně propojovacího
systému bezpečná a nesmí narušovat stanovenou funkční způsobilost in vitro diagnostika.
Každé omezení použití musí být uvedeno na značení popřípadě v návodu k použití.
3.2. In vitro diagnostika musí být navržena a vyrobena takovým způsobem,
aby byla nejvyšší možnou měrou minimalizována rizika spojená s jejich použitím za
přítomnosti materiálů, substancí a plynů, se kterými za normálních podmínek použití
mohou přijít do styku.
3.3. In vitro diagnostika musí být navržena a vyrobena takovým způsobem,
aby byla minimalizována
3.3.1. rizika poranění vyplývající z fyzikálních charakteristik in
vitro diagnostik (zejména prvky součinu objemu a tlaku, rozměru, a kde to přichází
v úvahu, i ergonomických vlastností),
3.3.2. rizika spojená s dostatečně předvídatelnými vnějšími vlivy,
jako jsou magnetická pole, vnější elektrické vlivy, elektrostatické výboje, tlak
a vlhkost vzduchu, teplota nebo změny tlaku nebo zrychlení nebo náhodné vniknutí
substancí do in vitro diagnostika. In vitro diagnostika musí být navržena a vyrobena
tak, aby poskytovala dostatečnou vnitřní odolnost proti elektromagnetickému rušení,
aby byla umožněna jejich funkce podle určení.
3.4. In vitro diagnostika musí být navržena a vyrobena tak, aby při normálním
provozu i při závadě byla co nejvíce snížena rizika požáru nebo výbuchu. Zvláštní
pozornost je nutno věnovat in vitro diagnostikům, při jejichž určeném použití se
vyskytují hořlavé látky nebo látky, které by mohly způsobit vznícení.
3.5. In vitro diagnostika musí být navržena a vyrobena tak, aby umožňovala
řízení bezpečné likvidace odpadu.
3.6. Stupnice pro měření, monitorování nebo zobrazování (včetně změny
barvy a jiných optických indikátorů) musí být navrženy a vyrobeny v souladu s ergonomickými
zásadami s přihlédnutím k určenému účelu in vitro diagnostika.
4.
In vitro diagnostika sloužící jako nástroj nebo přístroj s měřicí funkcí.
4.1. In vitro diagnostika, která jsou nástroji nebo přístroji s primární
analytickou měřicí funkcí, musí být navržena a vyrobena tak, aby poskytovala dostatečnou
stabilitu a přesnost měření v odpovídajících mezích přesnosti s přihlédnutím k určenému
účelu použití a s ohledem na dostupné a odpovídající metody měření a materiály. Meze
přesnosti musí být specifikovány výrobcem.
4.2. Jsou-li hodnoty vyjádřeny číselně, musí být uvedeny v zákonných jednotkách
v souladu s příslušnými předpisy.26)
5.
Ochrana před zářením.
5.1. In vitro diagnostika musí být navržena, vyrobena a zabalena tak, aby
se minimalizovalo vystavení uživatelů emitovanému záření.
5.2. V případě, že jsou in vitro diagnostika určena k emitování potenciálně
nebezpečného viditelného, popřípadě neviditelného záření, musí, pokud je to možné,
být
5.2.1. navržena a vyrobena tak, aby bylo zaručeno, že charakteristiky a množství
emitovaného záření lze řídit, popřípadě upravovat,
5.2.2. opatřena optickými displeji,
popřípadě zvukovými výstrahami o takových emisích.
5.3. Provozní instrukce pro in vitro diagnostika emitující záření musí
obsahovat podrobné informace o povaze emitovaného záření in vitro diagnostik, informace
o ochraně uživatele a o způsobech, jak zamezit zneužití a vyloučit rizika plynoucí
z instalace.
6.
Požadavky na in vitro diagnostika připojená ke zdroji energie nebo vybavená
zdrojem energie.
6.1. In vitro diagnostika obsahující elektronické programovatelné systémy,
včetně programového vybavení, musí být navržena tak, aby byla zajištěna opakovatelnost,
spolehlivost a funkční způsobilost těchto systémů podle určeného použití.
6.2. In vitro diagnostika musí být navržena a vyrobena tak, aby bylo sníženo
na minimum riziko vzniku elektromagnetického rušení, které by mohlo ovlivnit provoz
jiných in vitro diagnostik nebo zařízení v obvyklém prostředí.
6.3. In vitro diagnostika musí být navržena a vyrobena tak, aby za předpokladu
správné instalace bylo co možná nejvíce vyloučeno riziko náhodného úrazu elektrickým
proudem při normálním použití i při výskytu jedné závady.
6.4. Ochrana před mechanickými a tepelnými riziky.
6.4.1. In vitro diagnostika musí být navržena a vyrobena tak, aby byla
zaručena ochrana uživatele před mechanickými riziky. In vitro diagnostika musí být
za předvídaných podmínek provozu dostatečně stabilní. Musí být schopná odolávat namáhání
předpokládaného provozního prostředí a musí si zachovat tuto odolnost po dobu své
předpokládané životnosti, pokud jsou dodržovány veškeré požadavky týkající se kontroly
a údržby uvedené výrobcem. Kde existují rizika s ohledem na přítomnost pohyblivých
částí, rozlomení nebo odpojení nebo na únik látek, musí být začleněna příslušná ochranná
opatření.
Veškeré kryty nebo jiné ochranné mechanizmy zahrnuté v in vitro diagnostiku
za účelem ochrany, obzvláště před pohyblivými částmi, musí být bezpečné a nesmějí
bránit přístupu k in vitro diagnostiku při normálním provozu ani omezovat pravidelnou
údržbu tohoto prostředku podle určení výrobce.
6.4.2. In vitro diagnostika musí být navržena a vyrobena tak, aby se
maximálně snížilo riziko vibrací vyvolané těmito in vitro diagnostiky, a to s ohledem
na technický pokrok a dostupné možnosti k omezení těchto vibrací, zejména u jejich
zdroje, pokud tyto vibrace nejsou specifickou součástí funkční způsobilosti in vitro
diagnostik.
6.4.3. In vitro diagnostika musí být navržena a vyrobena tak, aby
se maximálně snížilo riziko vyplývající z hluku, který emitují, a to s ohledem na
technický pokrok a dostupné možnosti k omezení hluku, zejména u jejich zdroje, pokud
emitovaný hluk není specifickou součástí funkční způsobilosti in vitro diagnostika.
6.4.4. Koncové a připojovací části ke zdrojům elektrické energie, plynu
nebo hydraulické a pneumatické energie, se kterými musí uživatel manipulovat, musí
být navrženy a vyrobeny tak, aby byla minimalizována všechna možná rizika.
6.4.5. Přístupné části in vitro diagnostika (s výjimkou částí nebo
míst určených k dodávání tepla nebo k dosažení stanovených teplot) a jejich okolí
nesmějí dosahovat za normálních podmínek užití potenciálně nebezpečných teplot.
7.
Požadavky na in vitro diagnostika pro sebetestování.
In vitro diagnostika pro sebetestování musí být navržena a vyrobena tak,
aby jejich činnost odpovídala určenému účelu použití, s přihlédnutím k odbornosti
a dostupným prostředkům uživatelů a k vlivu způsobenému odchylkami, které lze očekávat
v uživatelských technických postupech a prostředí. Informace a instrukce poskytnuté
výrobcem by měly být pro uživatele lehce srozumitelné a snadno použitelné.
7.1. In vitro diagnostika pro sebetestování musí být navržena a vyrobena
tak, aby
7.1.1. byla pro uživatele snadno použitelné a
7.1.2. snižovala chybu uživatele
při zacházení s in vitro diagnostikem a při interpretaci výsledku.
7.2. In vitro diagnostika pro sebetestování musí, kde je to možné, zahrnovat
kontrolu uživatelem, tedy postup, kterým se uživatel může přesvědčit, že během doby
použití bude in vitro diagnostikum funkčně způsobilé v souladu s určeným účelem použití.
8.
Informace poskytované výrobcem.
8.1. Ke každému in vitro diagnostiku musí být poskytnuty informace potřebné
k jeho bezpečnému a řádnému použití, s přihlédnutím k proškolení a znalostem možných
uživatelů, a k identifikaci výrobce. Těmito informacemi se rozumějí údaje na značení
a v návodech k použití. Pokud je to proveditelné a vhodné, musí být informace potřebné
k bezpečnému použití in vitro diagnostika uvedeny na samotném in vitro diagnostiku,
popřípadě, kde to přichází v úvahu, na prodejním obalu. Pokud není úplné označování
každé jednotky in vitro diagnostika prakticky proveditelné, musí být informace uvedeny
na obalu, popřípadě v návodech k použití poskytnutých s jedním nebo více in vitro
diagnostiky.
Návod k použití musí být poskytnut uživateli nebo provázet in vitro
diagnostikum nebo být přiložen v balení jednoho nebo více in vitro diagnostik.
V
řádně zdůvodněných a výjimečných případech takovéto návody k použití nejsou pro in
vitro diagnostikum zapotřebí, jestliže může být řádně a bezpečně použit bez nich.
8.2. V případech, kde to přichází v úvahu, lze informace poskytnout ve
formě symbolů. Každý použitý symbol a identifikační barva musí být v souladu s harmonizovanými
normami. V oblastech, pro které harmonizované normy neexistují, musí být použité
symboly a barva popsány v průvodní dokumentaci k in vitro diagnostiku.
8.3. U in vitro diagnostik obsahujících látku, která může být považována
za nebezpečnou s ohledem na charakter a množství jejích složek a forem, ve kterých
jsou přítomny, musí být použity příslušné výstražné symboly a splněny požadavky na
označování podle příslušných předpisů.
V případech, kdy pro nedostatek místa celou
informaci nelze připojit na samotné in vitro diagnostikum nebo na jeho značení, umístí
se výstražné symboly na značení a ostatní informace požadované tímto nařízením se
uvedou v návodech k použití.
8.4. Značení musí obsahovat následující podrobné údaje, které podle vhodnosti
mohou být v podobě symbolů:
8.4.1. jméno, popřípadě jména a příjmení výrobce, adresu jeho bydliště
a místa (míst) podnikání, jestliže výrobcem je fyzická osoba; název nebo obchodní
firmu, adresu sídla, jestliže výrobcem je právnická osoba (dále jen "identifikační
údaje"). U in vitro diagnostik dovážených do členských států s předpokladem jejich
distribuce v České republice, musí značení, vnější obal nebo návody k použití obsahovat
i identifikační údaje zplnomocněného zástupce,
8.4.2. podrobné údaje nezbytné pro uživatele k jednoznačné identifikaci
in vitro diagnostika a obsahu obalu,
8.4.3. v případě potřeby slovo "STERILNÍ" ("STERILE") nebo prohlášení
ozřejmující jakýkoliv zvláštní mikrobiologický stav nebo stupeň čistoty,
8.4.4. kód šarže, před kterým je uveden symbol "ŠARŽE" ("LOT") nebo
sériové číslo,
8.4.5. v případě potřeby určení data, do kterého by in vitro diagnostikum
nebo jeho část měly být bezpečně použity bez zhoršení funkční způsobilosti vyjádřené
v tomto pořadí: rok, měsíc, a kde to přichází v úvahu, den,
8.4.6. v případě in vitro diagnostik pro ověření funkční způsobilosti,
slova "pouze pro ověření funkční způsobilosti",
8.4.7. v případě potřeby prohlášení udávající, že in vitro diagnostikum
je pro použití in vitro,
8.4.8. zvláštní skladovací, popřípadě manipulační podmínky,
8.4.9. v případě potřeby zvláštní provozní pokyny,
8.4.10. odpovídající výstrahy případně nutná bezpečnostní opatření,
8.4.11. pokud je in vitro diagnostikum určeno pro sebetestování, musí
být tato skutečnost zřetelně uvedena.
8.5. Není-li určený účel použití uživateli zřejmý, výrobce uvede tento
účel v návodech k použití a na značení in vitro diagnostiku, přichází-li to v úvahu.
8.6. Pokud je to účelné a prakticky proveditelné, musí být in vitro diagnostika
a oddělené součásti podle potřeby značeny údaji o šaržích, aby bylo možno učinit
odpovídající kroky ke zjištění rizik představovaných těmito in vitro diagnostiky
a jejich oddělitelnými součástmi.
8.7. V případě potřeby musí návod k použití obsahovat následující podrobné
údaje:
8.7.1. uvedené v bodu 8.4. s výjimkou bodu 8.4.4. a 8.4.5.,
8.7.2. o složení výsledku reakčních činidel (reakční produkt) podle
povahy a množství nebo koncentrace aktivní složky (složek) činidla (činidel) nebo
soupravy a vyjádření, kde to přichází v úvahu, že in vitro diagnostikum obsahuje
další složky, které mohou ovlivnit měření,
8.7.3. o podmínkách a době skladování po prvním otevření primární
nádoby, spolu se skladovacími podmínkami a stabilitou pracovních činidel,
8.7.4. o funkčních způsobilostech uvedených v části A bodu 3 této
přílohy,
8.7.5. týkající se určení veškerých zvláštních potřebných zařízení
včetně informací potřebných pro identifikaci takového zvláštního zařízení pro řádné
použití,
8.7.6. o typu vzorku plánovaného pro použití, o zvláštních podmínkách
odběru, postupu přípravy a v případě potřeby, skladovacích podmínkách, včetně instrukcí
pro přípravu pacienta,
8.7.7. o postupu, který je třeba dodržet při použití in vitro diagnostika,
8.7.8. o postupu měření, který je třeba dodržet u in vitro diagnostika,
a dále podle potřeby k:
8.7.8.1. podstatě postupu,
8.7.8.2. specifickým charakteristikám
analytické funkční způsobilosti (například citlivost, specificita, přesnost, opakovatelnost,
reprodukovatelnost, meze detekce a rozsah měření, včetně informací potřebných ke
zvládnutí známých významných rušivých vlivů), omezení metody a informace o použití
dostupných postupů a materiálů referenčního měření ze strany uživatele,
8.7.8.3.
dalším potřebným postupům nebo manipulaci před použitím in vitro diagnostika (například
rekonstituce, inkubace, ředění, kontrola nástrojů),
8.7.8.4. účelnosti požadavku
zvláštního školení,
8.7.9. týkající se matematického postupu, na jehož základě je proveden
výpočet analytického výsledku,
8.7.10. o opatřeních v případě změn analytické funkční způsobilosti
in vitro diagnostika,
8.7.11. týkající se vhodných informací pro uživatele o
8.7.11.1. vnitřní
kontrole jakosti včetně specifických validačních postupů,
8.7.11.2. zjistitelnosti
kalibrace in vitro diagnostika,
8.7.12. týkající se referenčních intervalů pro zjišťovaná množství,
včetně popisu příslušné referenční populace,
8.7.13. o vlastnostech in vitro diagnostika umožňujících identifikaci
vhodných in vitro diagnostik nebo vybavení, při jejichž použití je dosažena bezpečná
a řádná kombinace v případě, že in vitro diagnostikum musí být instalováno, spojeno
nebo použito v kombinaci s dalšími in vitro diagnostiky nebo vybavením, aby splňovalo
požadavky pro jeho určený účel použití,
8.7.14. potřebné k ověření, zda je in vitro diagnostikum řádně instalováno
a může správně a bezpečně pracovat, včetně potřebných podrobných údajů o povaze a
četnosti údržby a kalibraci, které jsou nezbytné k řádné a bezpečné funkci, informace
o bezpečné likvidaci odpadů,
8.7.15. o jakémkoli dalším potřebném zákroku nebo manipulaci před použitím
in vitro diagnostika (například sterilizace, konečná kompletace),
8.7.16. nezbytné pro případ poškození ochranného obalu a podrobné
údaje o vhodných resterilizačních nebo dekontaminačních postupech,
8.7.17. o vhodných postupech, které dovolují opakované použití in
vitro diagnostika včetně čištění, dezinfekce, balení, resterilizace, nebo dekontaminace
a omezení počtu opakovaných použití, jestliže je in vitro diagnostikum určeno k opakovanému
použití,
8.7.18. týkající se předběžných bezpečnostních opatření, která musí
být přijata za dostatečně předvídatelných podmínek prostředí z hlediska vystavení
účinkům magnetických polí, vnějších elektrických vlivů, elektrostatického výboje,
tlaků a tlakových změn, zrychlení, tepelných zdrojů vzplanutí, popřípadě dalších
okolností,
8.7.19. týkající se předběžných bezpečnostních opatření, která musí
být přijata proti jakýmkoli zvláštním a neobvyklým rizikům souvisejícím s použitím
nebo likvidací in vitro diagnostika, včetně zvláštních ochranných opatření, kde in
vitro diagnostikum obsahuje látky lidského nebo zvířecího původu, musí být věnována
pozornost jejich možné nakažlivé povaze,
8.7.20. o specifikacích pro in vitro diagnostika určená k sebetestování,
a to
8.7.20.1. výsledky musí být vyjadřovány a poskytovány způsobem, který je snadno
pochopitelný pro neodborníka; informace musí být poskytovány spolu s radou pro uživatele
a postupem, jde-li o pozitivní, negativní nebo nejasné výsledky, a o možnosti nesprávného
pozitivního nebo nesprávného negativního výsledku; zvláštní podrobné údaje mohou
být vynechány, pokud ostatní informace dodané výrobcem jsou dostatečné k tomu, aby
mohl uživatel použít in vitro diagnostikum a porozumět výsledku (výsledkům) získanému
(získaných) in vitro diagnostikem,
8.7.20.2. informace musí obsahovat vyjádření,
že uživatel by neměl činit jakékoliv závěry o zdravotním dopadu získaných výsledků,
aniž by tyto výsledky nejdříve konzultoval se svým lékařem,
8.7.20.3. informace
musí obsahovat poučení, je-li in vitro diagnostikum pro sebetestování použito pro
sledování existující nemoci, že pacient může pozměnit způsob léčby pouze tehdy, pokud
byl v tomto smyslu náležitě proškolen,
8.7.21. datum vydání poslední revize návodu k použití.
Příl.2
SEZNAM IN VITRO DIAGNOSTIK UVEDENÝCH V § 8 ODST. 2 a 3
Seznam A
1. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro stanovení krevních skupin:
systém AB0,
Rh (C, c, D, E, e) Kell(K),
2. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro průkaz, potvrzení a kvantifikaci ukazatelů HIV infekce (HIV 1 a 2),
HTLV I a II, a hepatitidy B, C a D v lidských vzorcích.
3. zkoušky na variantní Creutzfeldtovu-Jakobovu nemoc (vCJD) pro vyšetření
krve, diagnózu a potvrzení.
Seznam B
1. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro stanovení krevních skupin:
anti - Duffy, a
anti - Kidd,
2. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro stanovení:
nepravidelných protilátek proti erytrocytům,
3. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro zjištění a kvantifikaci v lidských vzorcích následujících vrozených
infekcí:
zarděnky, a
toxoplazmóza,
4. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro diagnózu následujících dědičných onemocnění:
fenylketonuria,
5. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro stanovení následujících lidských infekcí:
cytomegalovirus,
chlamydie,
6. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro stanovení HLA tkáňových skupin:
DR, A, B,
7. činidla a výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů pro stanovení následujících nádorových markerů:
PSA,
8. činidla s výsledky reakcí činidel, včetně příslušných kalibrátorů a kontrolních
materiálů a programového vybavení, navržených pro specifické vyhodnocení rizika:
trizómie 21,
9. následující in vitro diagnostikum pro sebetestování, včetně jeho příslušných
kalibrátorů a kontrolních materiálů:
in vitro diagnostikum k měření krevního cukru.
Příl.3
ES PROHLÁŠENÍ O SHODĚ
1. ES prohlášení o shodě je postup, kterým výrobce nebo jeho zplnomocněný
zástupce, plnící závazky podle bodu 2 až 5 a navíc v případě in vitro diagnostik
pro sebetestování podle bodu 6 této přílohy, zaručuje a prohlašuje, že dané výrobky
jsou v souladu s ustanoveními tohoto nařízení, která se na něj vztahují. Výrobce
opatřuje in vitro diagnostika označením CE v souladu s § 9.
2. Výrobce připraví technickou dokumentaci podle bodu 3 této přílohy a zajistí,
že výrobní postup odpovídá zásadám zabezpečování jakosti, vymezeným v bodu 4.
3. Technická dokumentace musí umožnit hodnocení shody in vitro diagnostika
s požadavky tohoto nařízení. Musí obsahovat zejména
3.1. všeobecný popis in vitro diagnostika včetně uvažovaných variant,
3.2. dokumentaci systému jakosti,
3.3. informace o konstrukci, včetně určení vlastností základních materiálů,
charakteristiky a limitace funkční způsobilosti in vitro diagnostik, výrobní technologie
a v případě přístrojů konstrukční výkresy, schéma součástí, podsestav a obvodů,
3.4. v případě in vitro diagnostik obsahujících tkáně lidského původu
nebo látek odvozených z takovýchto tkání, informace o původu takového materiálu a
o podmínkách, za kterých byly odebrány,
3.5. popis a vysvětlení nezbytné k porozumění výše uvedeným vlastnostem,
výkresům a diagramům a činnosti in vitro diagnostik,
3.6. výsledky analýzy rizika, a kde to přichází v úvahu, seznam plně
nebo částečně použitých norem podle § 4 a popisy řešení přijatých pro splnění základních
požadavků tohoto nařízení, pokud harmonizované normy podle § 4 nebyly použity v plném
rozsahu,
3.7. v případě sterilních in vitro diagnostik nebo in vitro diagnostik
se zvláštním mikrobiologickým stavem nebo stupněm čistoty, popis použitých postupů,
3.8. výsledky konstrukčních výpočtů a provedených kontrol,
3.9. má-li být in vitro diagnostikum spojeno s jiným in vitro diagnostikem
(in vitro diagnostika) za účelem dosažení určené funkce, musí být prokázáno, že při
spojení s jakýmkoliv takovým in vitro diagnostikem (in vitro diagnostiky) majícím
vlastnosti specifikované výrobcem, je ve shodě se základními požadavky,
3.10. zkušební záznamy,
3.11. dostatečné údaje o ověřování funkční způsobilosti, ukazující funkční
způsobilosti deklarované výrobcem a podložené referenčním systémem měření (je-li
k dispozici), spolu s informacemi o referenčních metodách, referenčních materiálech,
známých referenčních hodnotách, přesnosti a použitých jednotkách měření. Tyto údaje
by měly vycházet ze studií v klinickém nebo jiném odpovídajícím prostředí nebo vzejít
z odpovídajících biografických odkazů,
3.12. značení a návody k použití,
3.13. výsledky stabilitních studií.
4. Výrobce zajistí potřebnými opatřeními, aby výrobní postup zachovával zásady
zabezpečování jakosti odpovídající vyráběným in vitro diagnostikům.
Systém je zaměřen
na
4.1. organizační strukturu a odpovědnosti,
4.2. výrobní postupy a systematické
řízení jakosti výroby,
4.3. in vitro diagnostika ke sledování funkční způsobilosti
systému jakosti.
5. Výrobce zavede a udržuje aktualizovaný systematický postup pro vyhodnocování
zkušeností získaných s in vitro diagnostiky ve fázi po uvedení na trh a do provozu
a pro zavedení odpovídajících opatření umožňujících provedení jakýchkoliv potřebných
nápravných opatření se zřetelem k povaze a rizikům souvisejícím s in vitro diagnostikem.
Výrobce oznámí Ústavu každou potenciální možnost vzniku nežádoucí příhody nebo nežádoucí
příhodu, jakmile se o ní dozví.
6. U in vitro diagnostik pro sebetestování výrobce podá notifikované osobě
žádost o přezkoumání návrhu.
6.1. Žádost musí umožnit porozumění návrhu in vitro
diagnostiku a musí obsahovat dokumenty umožňující posouzení shody s požadavky tohoto
nařízení, vztahujícími se k návrhu. Žádost zahrnuje:
6.1.1. výsledky zkoušek, a kde
to přichází v úvahu, také výsledky studií provedených neodborníky,
6.1.2. údaje
dokumentující vhodnost zacházení s in vitro diagnostikem s ohledem na jeho určený
účel použití pro sebetestování,
6.1.3. informace, které jsou poskytovány s in vitro
diagnostikem na jeho značení a v návodech k použití.
6.2. Notifikovaná osoba prozkoumá
žádost, a pokud návrh odpovídá příslušným ustanovením tohoto nařízení, vydá žadateli
certifikát ES přezkoumání návrhu. Notifikovaná osoba může požadovat doplnění žádosti
dalšími zkouškami nebo důkazy za účelem posouzení shody s požadavky tohoto nařízení
vztahujícími se k návrhu.
Certifikát ES přezkoumání návrhu obsahuje závěry šetření,
podmínky platnosti, údaje potřebné k identifikaci schváleného návrhu, a kde to přichází
v úvahu, popis určeného účelu použití in vitro diagnostika.
6.3. Žadatel informuje
notifikovanou osobu, která vydala certifikát ES přezkoumání návrhu, o jakékoli podstatné
změně schváleného návrhu. Pokud by změny mohly ovlivnit shodu se základními požadavky
nebo by mohly ovlivnit předepsané podmínky použití in vitro diagnostika, potom změny
schváleného návrhu musí být dále schváleny notifikovanou osobou, která vydala certifikát
ES přezkoumání návrhu. Toto dodatečné schválení je vyhotoveno ve formě dodatku k
certifikátu ES přezkoumání návrhu.
Příl.4
ES PROHLÁŠENÍ O SHODĚ
(Systém úplného zabezpečování jakosti)
1.
Výrobce zajistí uplatnění systému jakosti schváleného pro návrh, výrobu a
výstupní kontrolu příslušných in vitro diagnostik podle bodu 3 této přílohy; výrobce
podléhá auditu uvedenému v bodu 3.3. a dozoru podle bodu 5.
U in vitro diagnostik
podle seznamu A přílohy č. 2 k tomuto nařízení výrobce navíc postupuje podle bodů
4 a 6.
ES prohlášení o shodě je postup, kterým výrobce, plnící závazky podle tohoto
bodu, zaručuje a prohlašuje, že příslušná in vitro diagnostika jsou v souladu s ustanoveními
tohoto nařízení, která se na ně vztahují.
2.
Výrobce
2.1. označuje in vitro diagnostika v souladu s § 9 a
2.2. vypracuje písemné prohlášení o shodě podle § 8.
3.
Systém jakosti
3.1. Výrobce
3.1.1. předkládá písemnou žádost notifikované osobě o posouzení (vyhodnocení
a schválení) svého systému jakosti. Žádost musí obsahovat:
3.1.1.1. jméno, popřípadě
jména a příjmení výrobce, adresu jeho bydliště a místa (míst) podnikání, jestliže
výrobcem je fyzická osoba; název nebo obchodní firmu, adresu sídla, jestliže výrobcem
je právnická osoba. V obou případech včetně adres výrobních míst, pro která platí
systém jakosti,
3.1.1.2. dostatečné informace o in vitro diagnostiku nebo kategorii
in vitro diagnostika, pro které postup platí,
3.1.1.3. písemné prohlášení, že u
žádné jiné notifikované osoby nebyla podána další žádost pro týž systém jakosti vztahující
se k in vitro diagnostiku,
3.1.1.4. dokumentaci systému jakosti,
3.1.1.5. závazek
výrobce plnit všechny povinnosti vyplývající ze schváleného systému jakosti,
3.1.1.6.
závazek výrobce udržovat systém jakosti v přiměřeném a účinném stavu,
3.1.1.7. závazek
výrobce zavést a aktualizovat systematický postup vyhodnocování zkušeností získaných
o in vitro diagnosticích v poprodejní fázi a pomocí vhodných in vitro diagnostik
učinit nezbytná nápravná opatření a oznámení v souladu s bodem 5 přílohy č. 3 k tomuto
nařízení.
3.2. Uplatňovaný systém jakosti musí zajistit, že in vitro diagnostika odpovídají
ustanovením, která se na ně vztahují z tohoto nařízení, a to v každém stadiu od jejich
návrhu až po výstupní kontrolu.
Všechny prvky, požadavky a opatření učiněná výrobcem
pro jím uplatňovaný systém jakosti musí být systematicky a řádně dokumentovány formou
písemně vypracovaných programů a postupů, a to programy jakosti, plány jakosti, příručkami
jakosti a záznamy o jakosti.
Dokumentace systému jakosti musí obsahovat zejména popis
3.2.1.
cílů jakosti výrobce,
3.2.2. organizace výrobce, zejména
3.2.2.1. organizačních struktur,
zodpovědnosti vedoucích pracovníků a jejich organizačních pravomocí týkajících se
jakosti návrhu a zhotovení in vitro diagnostika,
3.2.2.2. metod sledování chodu
systému jakosti a zejména jeho schopnosti dosáhnout požadované jakosti návrhu a in
vitro diagnostika, včetně kontroly těch in vitro diagnostik, které nejsou ve shodě,
3.2.3. postupů pro sledování a ověřování návrhu in vitro diagnostika, zejména
3.2.3.1.
všeobecný popis in vitro diagnostika včetně plánovaných variant,
3.2.3.2. veškerou
dokumentaci uvedenou v bodech 3.3. až 3.13. přílohy č. 3 k tomuto nařízení,
3.2.3.3.
v případě in vitro diagnostik pro sebetestování, informace uvedené v bodě 6.1. přílohy
č. 3 k tomuto nařízení,
3.2.3.4. použité techniky kontroly a ověřování návrhu, postupů
a systematických opatření, používaných při navrhování in vitro diagnostik,
3.2.4.
techniky kontrol a zabezpečování jakosti ve stadiu výroby, zejména:
3.2.4.1. metody
a postupy, které budou použity, zejména s ohledem na sterilizaci,
3.2.4.2. postupy
vztahující se k nákupu,
3.2.4.3. postupy k identifikaci výrobku, vypracované a uchovávané
v aktualizovaném stavu v každém stadiu výroby na základě výkresů, specifikací a dalších
souvisejících dokumentů,
3.2.5. příslušných testů a zkoušek, které budou provedeny
před výrobou, během výroby a po výrobě, jejich četnosti a použitých zkušebních zařízení,
musí být možné přiměřeným způsobem zpětně zjistit správnost kalibrace.
Výrobce provádí
požadovaná kontrolní šetření a zkoušky podle nejnovějšího stavu vědy a techniky.
Kontrolní šetření a zkoušky zahrnou výrobní postup včetně popisu surovin a jednotlivých
in vitro diagnostik nebo každé šarže vyrobených těchto in vitro diagnostik. Při zkoušení
in vitro diagnostik uvedených v seznamu A přílohy č. 2 k tomuto nařízení, zohlední
výrobce nejnovější dostupné informace, zejména s ohledem na biologickou komplexnost
a variabilitu vzorků, které mají být zkoušeny daným in vitro diagnostikem.
3.3. Notifikovaná osoba provádí audity systému jakosti, aby zjistila, zda
systém splňuje požadavky uvedené v bodu 3.2. této přílohy. Shoda s těmito požadavky
se předpokládá u systémů jakosti, které používají odpovídající harmonizované normy.
Tým provádějící hodnocení musí mít zkušenosti s hodnocením příslušných technologií.
Postup hodnocení zahrnuje prohlídku provozních prostor výrobce a v oprávněných případech
i kontrolu výrobních postupů v provozních prostorech dodavatelů, popřípadě dalších
smluvních stran výrobce.
Notifikovaná osoba po provedeném auditu systému jakosti oznámí
výrobci rozhodnutí, které musí obsahovat závěry kontroly a odůvodnění zhodnocení.
3.4. Výrobce informuje notifikovanou osobu, která schválila systém jakosti,
o každém záměru, který podstatně mění systém jakosti nebo jím pokrytý okruh in vitro
diagnostik. Notifikovaná osoba zhodnotí navrhované změny, ověří, zda takto změněný
systém jakosti ještě vyhovuje požadavkům uvedeným v bodu 3.2. této přílohy a oznámí
své rozhodnutí výrobci. Toto rozhodnutí musí obsahovat závěry kontroly a odůvodněné
zhodnocení.
4.
Přezkoumání návrhu in vitro diagnostik.
4.1. U in vitro diagnostik podle seznamu A přílohy č. 2 k tomuto nařízení,
navíc k závazkům výrobce podle bodu 3 této přílohy podává výrobce notifikované osobě
žádost o přezkoumání návrhu vztahujícího se k in vitro diagnostiku, které zamýšlí
výrobce vyrábět, a které je zařazeno do kategorie uvedené v bodu 3.1. této přílohy.
4.2. Žádost musí popisovat návrh, výrobu a obsahovat údaje o funkční způsobilosti
daného in vitro diagnostika. Musí obsahovat dokumenty, které umožní vyhodnotit, zda
in vitro diagnostikum splňuje požadavky podle bodu 3.2.3. této přílohy.
4.3. Notifikovaná osoba
4.3.1. přezkoumá žádost a pokud in vitro diagnostikum vyhovuje příslušným
ustanovením tohoto nařízení vydá žadateli certifikát ES přezkoumání návrhu in vitro
diagnostika,
4.3.2. si může vyžádat doplnění žádosti dalšími zkouškami nebo důkazy,
které umožní posoudit shodu in vitro diagnostika s požadavky tohoto nařízení,
4.3.3. uvede v certifikátu ES přezkoumání návrhu závěry přezkoumání,
podmínky platnosti, údaje potřebné k identifikaci schváleného návrhu a v případě
potřeby popis určeného účelu použití in vitro diagnostika.
4.4. Změny schváleného návrhu, které by mohly ovlivnit shodu se základními
požadavky tohoto nařízení nebo podmínky předepsané pro použití in vitro diagnostika,
podléhají dodatečnému schválení notifikovanou osobou, která vydala certifikát ES
přezkoumání návrhu. Žadatel informuje notifikovanou osobu, která vydala certifikát
ES přezkoumání návrhu, o všech změnách schváleného návrhu. Toto doplňkové schválení
má podobu dodatku k certifikátu ES přezkoumání návrhu.
4.5. Výrobce bezodkladně informuje notifikovanou osobu o zjištěných údajích
vztahujících se ke změnám patogenů a markerů infekčních nákaz, které se zkoumají,
obzvláště, pokud došlo ke změnám následkem biologické komplexnosti a variability.
V této souvislosti výrobce informuje notifikovanou osobu o tom, zda by některá z
těchto změn mohla ovlivnit funkční způsobilost daného in vitro diagnostika.
5.
Dozor
5.1. Cílem dozoru je zajistit, aby výrobce náležitě plnil závazky vyplývající
ze schváleného systému jakosti.
5.2. Výrobce zmocňuje notifikovanou osobu k provádění všech nezbytných
prohlídek a poskytne ji všechny příslušné informace, zejména
5.2.1. dokumentaci systému jakosti,
5.2.2. údaje stanovené systémem jakosti v oblasti návrhu, především výsledky
analýz, propočtů, zkoušek,
5.2.3. údaje stanovené systémem jakosti pro oblast výroby, především
zprávy z kontrol a data ze zkoušení, údaje o kalibraci a zprávy o kvalifikaci příslušných
pracovníků.
5.3. Notifikovaná osoba
5.3.1. provádí periodicky příslušné prohlídky a hodnocení
tak, aby se ujistila, že výrobce používá schválený systém jakosti a
5.3.2. poskytuje
výrobci hodnotící zprávu,
5.3.3. provádí, podle svého uvážení, u výrobce i předem
neohlášené kontroly, při nichž je oprávněna v případě potřeby provést nebo si vyžádat
provedení zkoušky za účelem kontroly, zda systém jakosti je řádně účinný.
5.3.4.
poskytuje výrobci zprávu o provedené kontrole, popřípadě i výsledku zkoušky, jestliže
byla provedena.
6.
Ověřování vyrobených in vitro diagnostik uvedených v seznamu A přílohy č.
2 k tomuto nařízení.
6.1. V případě in vitro diagnostik uvedených v seznamu A přílohy č. 2 k
tomuto nařízení výrobce bezodkladně poskytne notifikované osobě po skončení kontrolních
šetření a zkoušek příslušné záznamy o zkouškách provedených na vyrobených in vitro
diagnostikách nebo na každé výrobní šarži těchto in vitro diagnostik.
Dále výrobce
poskytne notifikované osobě vzorky vyrobených in vitro diagnosticích nebo výrobních
šarží in vitro diagnostik v souladu s předem dohodnutými podmínkami a postupy.
6.2. Výrobce může uvést in vitro diagnostika na trh, pokud notifikovaná
osoba v dohodnuté časové lhůtě ne delší než 30 dnů po převzetí vzorků nesdělí výrobci
jiné rozhodnutí obsahující zejména jakoukoliv podmínku platnosti vydaných certifikátů.
Příl.5
ES PŘEZKOUŠENÍ TYPU
1. ES přezkoušení typu in vitro diagnostika je součástí postupu, kterým notifikovaná
osoba zjišťuje a osvědčuje, že reprezentativní vzorek posuzované výroby splňuje příslušná
ustanovení tohoto nařízení.
2. Žádost o ES přezkoušení typu podává notifikované osobě výrobce nebo jeho
zplnomocněný zástupce. Žádost musí obsahovat
2.1. jméno, popřípadě jména a příjmení
výrobce, adresu jeho bydliště a místa (míst) podnikání, jestliže výrobcem je fyzická
osoba; název nebo obchodní firmu, adresu sídla, jestliže výrobcem je právnická osoba,
2.2. jméno, příjmení a adresu trvalého pobytu, jestliže podává žádost zplnomocněný
zástupce, který je fyzickou osobou, nebo název (obchodní firmu) a adresu sídla, podává-li
žádost zplnomocněný zástupce, který je právnickou osobou,
2.3. dokumentaci podle
bodu 3 této přílohy, která je potřebná k posouzení shody typu s požadavky tohoto
nařízení. Žadatel předá vzorek in vitro diagnostika notifikované osobě, která si
podle svého uvážení vyžádá další vzorky.
2.4. písemné prohlášení, že stejná žádost
nebyla podána žádné jiné notifikované osobě.
3. Dokumentace musí umožnit pochopení návrhu, výroby a funkční způsobilosti
in vitro diagnostika. Dokumentace obsahuje zejména:
3.1. všeobecný popis typu, včetně
zamýšlených variant,
3.2. dokumentaci uvedenou v bodech 3.3. až 3.13. přílohy č.
3 k tomuto nařízení,
3.3. v případě in vitro diagnostik pro sebetestování informace
uvedené v bodě 6.1. přílohy č. 3 k tomuto nařízení.
4. Notifikovaná osoba
4.1. přezkoumá a zhodnotí dokumentaci a ověří, zda
byl vzorek in vitro diagnostika vyroben v souladu s dokumentací, a zaznamenává všechny
položky, které jsou navrženy v souladu s příslušnými harmonizovanými normami podle
§ 4, podobně jako položky, které nejsou navrženy podle těchto norem,
4.2. provede
nebo nechá provést příslušné kontroly a zkoušky, nezbytné k ověření, zda řešení přijatá
výrobcem splňují základní požadavky tohoto nařízení, a to v případě nepoužití norem
podle § 4. Jestliže in vitro diagnostikum má být spojeno s jiným in vitro diagnostikem,
popřípadě s jinými in vitro diagnostiky za účelem dosažení jeho určené funkce, musí
být prokázáno, že při spojení s jakýmkoliv takovým in vitro diagnostikem, popřípadě
in vitro diagnostiky s vlastnostmi specifikovanými výrobcem, je ve shodě se základními
požadavky.
4.3. provede nebo nechá provést příslušné kontroly nebo zkoušky nezbytné
k ověření, zda výrobce skutečně použil odpovídající harmonizované normy, které zvolil,
4.4. dohodne se žadatelem místo, kde budou nezbytné kontroly a zkoušky prováděny.
5. Splňuje-li vzorek in vitro diagnostika ustanovení tohoto nařízení, notifikovaná
osoba vydá žadateli certifikát ES přezkoušení typu. Certifikát ES přezkoušení typu
musí obsahovat
5.1. jméno, popřípadě jména a příjmení výrobce, adresu jeho bydliště
a místa (míst) podnikání, jestliže výrobcem je fyzická osoba; název nebo obchodní
firmu, adresu sídla, jestliže výrobcem je právnická osoba,
5.2. závěry kontroly,
podmínky platnosti a údaje potřebné k identifikaci schváleného typu. Příslušné části
dokumentace musí být připojeny k certifikátu ES přezkoušení typu; kopii certifikátu
ES přezkoušení typu a příslušné části dokumentace připojené k certifikátu ES přezkoušení
typu uchovává notifikovaná osoba.
6. Výrobce bezodkladně informuje notifikovanou osobu, která vydala certifikát
ES přezkoušení typu, o zjištěných údajích vztahujících se ke změnám patogenů a markerů
nákaz, které se zkoumají, zejména pokud došlo k těmto změnám následkem biologické
komplexnosti a variability. V této souvislosti výrobce informuje notifikovanou osobu
o tom, zda by některá z těchto změn mohla ovlivnit funkční způsobilost in vitro diagnostika.
6.1. Pro změny schváleného in vitro diagnostika je nutný dodatečný souhlas notifikované
osoby, která certifikát ES přezkoušení typu vydala, a to v případě, kdy změny mohou
ovlivnit shodu se základními požadavky tohoto nařízení nebo s podmínkami stanovenými
pro použití in vitro diagnostika. Žadatel informuje notifikovanou osobu, která vydala
certifikát ES přezkoušení typu, o každé významné změně provedené na schváleném in
vitro diagnostiku. Toto doplňkové schválení má podobu dodatku k původnímu certifikátu
ES přezkoušení typu.
7. Administrativní opatření Notifikované osoby mohou obdržet kopie certifikátů
ES přezkoušení typu, popřípadě jejich dodatků. Přílohy certifikátů ES přezkoušení
typu musí být dostupné jiným notifikovaným osobám na základě zdůvodněné žádosti a
po předchozím informování výrobce.
Příl.6
ES OVĚŘOVÁNÍ
1.
ES ověřování je postup, kterým výrobce nebo jeho zplnomocněný zástupce zajišťuje
a prohlašuje, že výrobky, které byly podrobeny postupu podle bodu 4 této přílohy,
odpovídají typu popsanému v certifikátu ES přezkoušení typu a vyhovují ustanovením
tohoto nařízení.
2.
Postup výrobce.
Výrobce
2.1. Výrobce provede nezbytná opatření, aby zajistil, že vyráběná in vitro
diagnostika odpovídají typu uvedenému v certifikátu ES přezkoušení typu a vyhovují
požadavkům, které se na ně vztahují z tohoto nařízení,
2.1.1. před zahájením výroby
výrobce připraví
2.1.1.1. výrobní proces zejména pokud jde o sterilizaci, jestliže
je to nutné,
2.1.1.2. rutinní postupy s předběžnými opatřeními, která mají být zavedena
k zajištění stejnorodosti výroby,
2.1.1.3. podle potřeby i zajištění souladu in
vitro diagnostik s typem popsaným v certifikátu ES přezkoušení typu a s požadavky,
které se na ně vztahují z tohoto nařízení.
2.2. Vzhledem k tomu, že některé podmínky konečného zkoušení podle bodu 6.3.
této přílohy nejsou vhodné, zavede výrobce se souhlasem notifikované osoby přiměřené
metody zkoušení, monitorování a kontroly používaných postupů. Ustanovení bodu 5 přílohy
č. 4 k tomuto nařízení platí ve vztahu k výše uvedeným schváleným postupům.
3.
Výrobce zavede a udržuje aktualizovaný systematický postup pro vyhodnocování
zkušeností získaných s in vitro diagnostiky v poprodejní fázi a pro uplatnění odpovídajících
opatření podle bodu 5 přílohy č. 3 k tomuto nařízení.
4.
Notifikovaná osoba.
4.1. Notifikovaná osoba provede odpovídající přezkoumání a zkoušky podle
bodu 2.2. této přílohy k ověření shody in vitro diagnostika s požadavky tohoto nařízení
podle rozhodnutí výrobce u
4.1.1. každého in vitro diagnostika podle bodu 5 této
přílohy nebo
4.1.2. vybraných statisticky podle bodu 6 této přílohy. Při statistickém
ověřování podle bodu 6 této přílohy notifikovaná osoba musí rozhodnout, zda musí
být použity statistické postupy pro průběžnou kontrolu šarží, nebo pro kontrolu samostatné
šarže. Toto rozhodnutí musí být učiněno po projednání s výrobcem. Pokud provedení
přezkoumání a zkoušek na statistickém podkladě nepřipadá v úvahu, přezkoumání a zkoušky
mohou být provedeny náhodně za předpokladu, že takovýto postup společně s opatřeními
přijatými v souladu s bodem 2.2. této přílohy zajišťuje rovnocennou úroveň shody.
5.
Ověřování přezkoumáním a zkouškami každého in vitro diagnostika.
Notifikovaná osoba
5.1. zkouší každé in vitro diagnostikum individuálně, za účelem ověření
shody každého in vitro diagnostika a s typem popsaným v certifikátu ES přezkoušení
typu s požadavky, které se na ně vztahují z tohoto nařízení. Notifikovaná osoba provádí
odpovídající zkoušky stanovené v příslušné harmonizované normě (harmonizovaných normách)
v souladu s § 4 a v případě potřeby provádí ekvivalentní zkoušky,
5.2. umístí nebo nechá umístit na každé schválené in vitro diagnostikum
své identifikační číslo a vypracuje písemný certifikát shody vztahující se k provedeným
zkouškám.
6.
Statistické ověřování
6.1. výrobce předloží vyrobená in vitro diagnostika ve stejnorodých výrobních
dávkách (šaržích),
6.2. notifikovaná osoba odebere z každé výrobní dávky (šarže) jeden nebo
více náhodně vybraných vzorků. In vitro diagnostika, která tvoří tento vzorek, se
zkoumají a podrobí se příslušným zkouškám stanoveným v odpovídající harmonizované
normě, popřípadě (harmonizovaných normách) podle § 4 nebo ekvivalentním zkouškám,
aby byla ověřena shoda in vitro diagnostik s typem popsaným v certifikátu ES přezkoušení
typu a s požadavky tohoto nařízení, které se na ně vztahují, aby mohlo být rozhodnuto
o přijetí výrobní dávky (šarže).
6.3. Statistická kontrola in vitro diagnostik vychází z vlastností, popřípadě
variant odběrových schémat s provozními vlastnostmi, které zajišťují vysokou úroveň
bezpečnosti a funkční způsobilosti podle současného stavu vědy a techniky. Plán odběru
vzorků se stanoví v souladu s harmonizovanými normami podle ustanovení § 4 a se zřetelem
ke specifické povaze příslušných in vitro diagnostik.
6.4. Jestliže notifikovaná osoba výrobní dávku (šarži) převezme, umístí
nebo nechá umístit na každé in vitro diagnostikum své identifikační číslo a vypracuje
písemný certifikát o shodě s odvoláním na provedené zkoušky. Všechna in vitro diagnostika
z výrobní dávky (šarže) mohou být uvedena na trh s výjimkou těch, jež nevyhověla.
Jestliže notifikovaná osoba vyhodnotí výrobní dávku (šarži) jako nevyhovující, učiní
přiměřená opatření, aby zabránila uvedení šarže na trh. V případě opakovaného odmítnutí
převzetí výrobní dávky (šarže) může notifikovaná osoba statistické ověřování pozastavit.
Výrobce může na zodpovědnost notifikované osoby připojit na in vitro diagnostikum
jeho identifikační číslo již v průběhu výrobního procesu.
Příl.7
ES PROHLÁŠENÍ SHODĚ
(Zabezpečení jakosti výroby)
1.
Výrobce zajišťuje uplatňování systému jakosti, schváleného pro výrobu daných
in vitro diagnostik, a provádí výstupní kontrolu podle bodu 3 této přílohy.
Výrobce
podléhá dozoru podle bodu 4 této přílohy.
ES prohlášení o shodě je součástí postupu,
kterým výrobce, plnící závazky podle tohoto bodu, zajišťuje a prohlašuje, že příslušná
in vitro diagnostika odpovídají typu popsanému v certifikátu ES přezkoušení typu
a že jsou v souladu s ustanoveními tohoto nařízení, které se na ně vztahují.
2.
Výrobce
2.1. opatřuje in vitro diagnostika označením CE v souladu s § 9 a
2.2. vypracuje prohlášení o shodě.
3.
Systém jakosti.
3.1. Výrobce předkládá notifikované osobě žádost o zhodnocení svého systému
jakosti. Žádost musí obsahovat
3.1.1. jméno, popřípadě jména a příjmení výrobce,
adresu jeho bydliště a místa (míst) podnikání, jestliže výrobcem je fyzická osoba;
název nebo obchodní firmu, adresu sídla, jestliže výrobcem je právnická osoba,
3.1.2.
dokumentaci a závazky uvedené v bodě 3.1. přílohy č. 4 k tomuto nařízení a
3.1.3.
technickou dokumentaci vztahující se ke schváleným typům a kopii certifikátu ES přezkoušení
typu.
3.2. Uplatnění systému jakosti musí zajistit, že vyrobená in vitro diagnostika
odpovídají typu popsanému v certifikátu ES přezkoušení typu. Všechny prvky, požadavky
a postupy přijaté výrobcem pro jeho systém jakosti musí být dokumentovány systematickým
a uspořádaným způsobem ve formě písemných vyjádření zásad a postupů. Tato dokumentace
systému jakosti musí umožnit jednotný výklad zásad jakosti a postupů, jako jsou programy
jakosti, plány, příručky a záznamy.
Dokumentace musí obsahovat zejména odpovídající
popis
3.2.1. cílů jakosti u výrobce,
3.2.2. organizace výrobce, zejména
3.2.2.1. organizačních
struktur, zodpovědnosti vedoucích zaměstnanců jejich pravomocí ve vztahu k jakosti
návrhu a výrobě in vitro diagnostika,
3.2.2.2. metod sledování účinnosti systému
jakosti a zejména jeho schopnosti dosáhnout požadované jakosti návrhu a in vitro
diagnostika, včetně kontroly in vitro diagnostik, které požadované jakosti nedosáhnou,
3.2.3. techniky kontrol a zajištění jakosti ve stadiu výroby, zejména
3.2.3.1.
postupy a procesy, které budou použity, zejména pokud jde o sterilizaci,
3.2.3.2.
postupy hodnocení dodavatelů,
3.2.3.3. postupy identifikace in vitro diagnostika
vyhotovené, uchovávané a aktualizované ve všech stadiích výroby na základě výkresů,
specifikací a dalších souvisejících dokumentů,
3.2.4. příslušné testy a zkoušky,
které budou používány před výrobou, během výroby a po výrobě, jejich četnost provádění
a použitá zkušební zařízení. Zpětně musí být možné přiměřeným způsobem vysledovat
správnost kalibrace zkušebních zařízení.
3.3. Notifikovaná osoba
3.3.1. provádí audity systému jakosti výroby, za
účelem zjištění, zda systém jakosti výroby vyhovuje požadavkům podle bodu 3.2. této
přílohy. Shoda s těmito požadavky se předpokládá u systémů jakosti výroby, ve kterých
se používají odpovídající harmonizované normy.
3.3.2. Tým provádějící hodnocení
systému jakosti výroby musí mít zkušenosti s hodnoceními příslušné technologie. Postup
hodnocení zahrnuje prohlídku provozních prostor výrobce a v oprávněných případech
i kontrolu výrobních postupů v provozních prostorech dodavatelů, popřípadě subdodavatelů
výrobce.
3.3.3. Po provedení auditu systému jakosti výroby notifikovaná osoba oznámí
výrobci nebo jeho zmocněnému zástupci rozhodnutí, které musí obsahovat závěry kontroly
a odůvodněné zhodnocení.
3.4. Výrobce informuje notifikovanou osobu, která schválila systém jakosti
výroby, o záměru podstatně změnit tento systém.
Notifikovaná osoba zhodnotí navrhované
změny a ověří, zda takto změněný systém jakosti výroby ještě vyhovuje požadavkům
podle bodu 3.2. této přílohy. Výrobci oznámí své rozhodnutí, které musí obsahovat
závěry kontroly a odůvodněné zhodnocení.
4.
Dozor.
Při dozoru se postupuje podle bodu 5 přílohy č. 4 k tomuto nařízení.
5.
Ověřování vyrobených in vitro diagnostik uvedených v seznamu A přílohy č.
2 k tomuto nařízení.
5.1. V případě in vitro diagnostik uvedených v seznamu A přílohy č. 2 k
tomuto nařízení, výrobce bezodkladně poskytne notifikované osobě po uzavření kontrolních
šetření příslušné záznamy o zkouškách provedených na vyrobených in vitro diagnosticích
nebo na každé výrobní dávce (šarži) in vitro diagnostik. Dále výrobce poskytne notifikované
osobě vzorky vyrobených in vitro diagnostik nebo výrobních dávek (šarží) in vitro
diagnostik v souladu s předem dohodnutými podmínkami a postupy.
5.2. Výrobce může uvést in vitro diagnostika na trh, pokud notifikovaná
osoba v dohodnuté časové lhůtě, ale ne delší než 30 dní po převzetí vzorků, nesdělí
výrobci jiné rozhodnutí, obsahující zejména jakoukoliv podmínku platnosti vydaných
certifikátů.
Příl.8
PROHLÁŠENÍ A POSTUPY TÝKAJÍCÍ SE IN VITRO DIAGNOSTIK PRO HODNOCENÍ FUNKČNÍ
ZPŮSOBILOSTI
1.
Výrobce nebo jeho zplnomocněný zástupce vyhotoví k in vitro diagnostikům
pro hodnocení funkční způsobilosti prohlášení obsahující údaje podle bodu 2 této
přílohy a zajistí splnění příslušných ustanovení podle tohoto nařízení.
2.
Prohlášení musí obsahovat
2.1. údaje umožňující identifikaci příslušných in vitro diagnostik,
2.2. plán hodnocení uvádějící zejména účel, vědecké a technické nebo zdravotnické
důvody, rozsah hodnocení a počet in vitro diagnostik, jichž se týká,
2.3. seznam laboratoří nebo jiných osob podílejících se na hodnotící studii,
2.4. datum zahájení a plánované trvání hodnocení a místo a počet zúčastněných
subjektů hodnocení (neodborníků) v případě in vitro diagnostik pro sebetestování,
2.5. prohlášení, že dotyčné in vitro diagnostikum je ve shodě s požadavky
tohoto nařízení, kromě hodnocených aspektů a aspektů specificky uvedených v prohlášení,
a že byla učiněna všechna předběžná opatření k ochraně zdraví a bezpečnosti uživatelů.
3.
Výrobce zpřístupňuje příslušným úřadům dokumentaci umožňující porozumět
návrhu, výrobě a funkční způsobilosti in vitro diagnostika, včetně očekávané funkční
způsobilosti, aby bylo možné zhodnocení souladu in vitro diagnostik s požadavky tohoto
nařízení. Tato dokumentace musí být uchovávána po dobu alespoň 5 let po skončení
hodnocení funkční způsobilosti.
Výrobce provádí nezbytná opatření nutná k tomu,
aby výrobní proces zajišťoval, že vyrobená in vitro diagnostika jsou ve shodě s dokumentací
zmíněnou v prvním odstavci.
4.
Ustanovení o oznámení podle § 11 se vztahuje na in vitro diagnostika určená
pro hodnocení funkční způsobilosti.
Příl.9
ZÁKLADNÍ KRITÉRIA PRO NOTIFIKOVANÉ OSOBY
Pro udělení autorizace se použijí následující kritéria:
1. notifikovaná osoba a osoby, které jsou členy jejích statutárních či jiných
orgánů, zaměstnanci notifikované osoby, popřípadě osoby, které na smluvním základě
jsou zodpovědní za hodnotící a ověřovací činnosti uvedené v přílohách č. 3 až 7 k
tomuto nařízení (dále jen "hodnocení a ověřování"), nesmějí
1.1. být konstruktérem,
výrobcem, dodavatelem ani servisním pracovníkem nebo uživatelem hodnocených a ověřovaných
in vitro diagnostik,
1.2. být ani zmocněným zástupcem osoby uvedené v bodu 1 této
přílohy,
1.3. podílet se přímo na návrhu, konstrukci, prodeji a údržbě in vitro
diagnostik ani zastupovat strany zúčastněné na těchto činnostech.
Možnost výměny
technických informací mezi výrobcem a notifikovanou osobou není předchozí částí bodu
1 této přílohy, dotčena,
2. notifikovaná osoba, její zaměstnanci, popřípadě osoby, které na smluvním
základě provádějí hodnocení a ověřování s nejvyšším stupněm profesionální péče a
potřebné způsobilosti v oblasti in vitro diagnostik, nesmějí být pod vlivem jakýchkoliv
nátlaků a stimulů, zejména finančních, které by mohly ovlivnit jejich úsudek nebo
výsledky kontrolní činnosti, zejména pochází-li tyto od osob nebo skupin osob, které
mají zájem na výsledcích hodnocení a ověřování,
3. notifikovaná osoba
3.1. při smluvním zajišťování specifických úkolů spojených
s hodnocením a ověřováním skutečností se ujistí, že subdodavatel vyhovuje ustanovením
tohoto nařízení,
3.2. uchová pro potřebu příslušných úřadů nebo orgánů potřebné
dokumenty, které hodnotí kvalifikaci subdodavatele a práci, kterou vykonal podle
tohoto nařízení,
3.3. je schopna vykonávat všechny úkoly, které jsou jí určeny podle
jedné z příloh č. 3 až 7 k tomuto nařízení, a pro které byla ustanovena, a to buď
sama, nebo je dát provést na svou odpovědnost,
3.4. má k dispozici nezbytný personál
a vlastní potřebná vybavení, která jí umožní řádně plnit technické i administrativní
úkoly uložené pro hodnocení a ověřování. Jedná se zejména o dostupnost dostatečného
vědeckého personálního zázemí s dostatečnými zkušenostmi a znalostmi potřebnými k
hodnocení biologické a funkční způsobilosti in vitro diagnostik, pro která byla autorizována,
s ohledem na požadavky tohoto nařízení a zejména na požadavky přílohy č. 1 k tomuto
nařízení,
3.5. má přístup k zařízením potřebným pro požadovaná ověření,
4. zaměstnanci notifikované osoby, popřípadě osoby, které na smluvním základě
jsou odpovědní za hodnocení a ověřování musí mít
4.1. řádnou odbornou kvalifikaci
ve všech hodnotících a ověřovacích postupech, v rámci a rozsahu udělené autorizace,
4.2. uspokojivé znalosti pravidel kontrolních postupů, které provádějí, a přiměřené
zkušenosti s touto činností,
4.3. schopnosti potřebné k vyhotovování certifikátů,
záznamů a zpráv prokazujících, že kontrolní postupy byly provedeny,
5. odměny zaměstnanců notifikované osoby a popřípadě osob, které na smluvním
základě odpovídají za hodnocení a ověřování nesmějí být závislé na počtu provedených
kontrol ani jejich výsledcích,
6. povinnost notifikované osoby uzavřít smlouvu o pojištění odpovědnosti
za škodu stanoví zvláštní právní předpis,27)
7. povinnost osob uvedených v této příloze, zachovávat mlčenlivost o skutečnostech,
o nichž se dozvídají při činnostech notifikované osoby stanoví zvláštní právní předpis.28)
Příl.11
nadpis vypuštěn
1.
Formulář pro oznámení osoby nakládající s in vitro diagnostiky uvedené
v § 11 odst. 1 nařízení vlády č. 453/2004 Sb., ve znění nařízení vlády č. 67/2011
Sb.
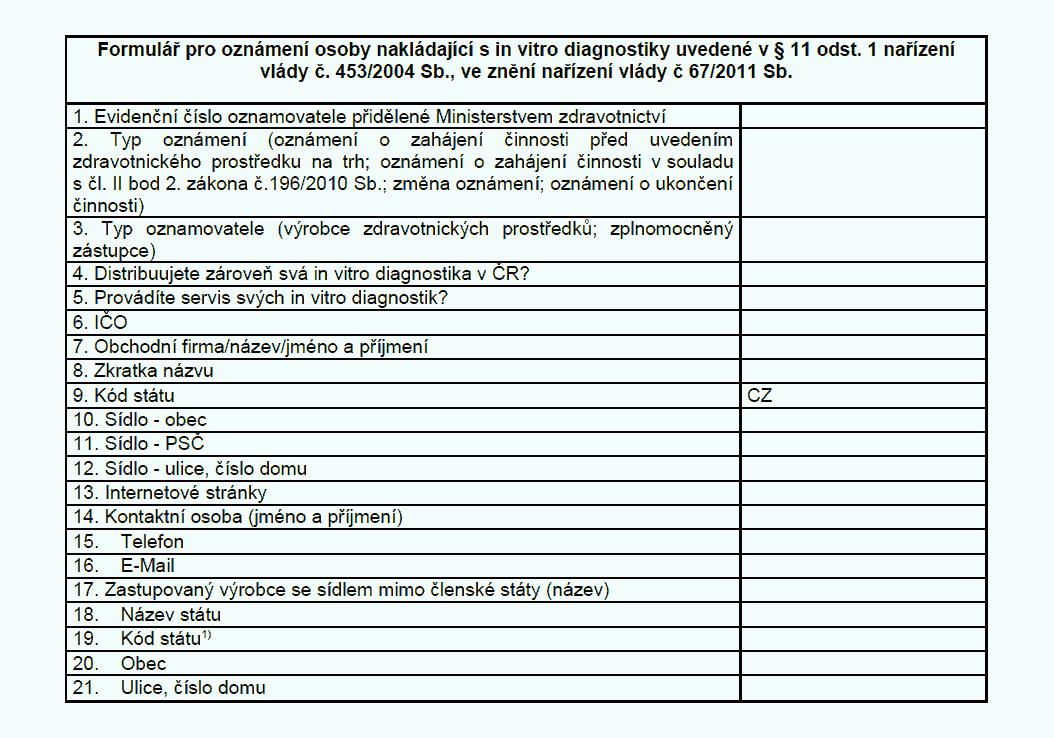
1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
AT Rakousko,
BE Belgie,
BG Bulharsko,
CH Švýcarsko,
CY Kypr,
CZ Česká republika,
DE Německo,
DK Dánsko,
EE Estonsko,
ES Španělsko,
FI Finsko,
FR Francie,
GB Spojené království,
GR Řecko,
HU Maďarsko,
IE Irsko,
IS Island,
IT Itálie,
LI Lichtenštejnsko,
LT Litva,
LU Lucembursko,
LV Lotyšsko,
MT Malta,
NL Nizozemsko,
NO Norsko,
PL Polsko,
PT Portugalsko,
RO Rumunsko,
SE Švédsko,
SI Slovinsko,
SK Slovensko.
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1.
a způsob jeho vyplnění zveřejní Ministerstvo zdravotnictví na svých internetových
stránkách.
2.2. Zplnomocněný zástupce podle § 3 písm. l) zákona o zdravotnických
prostředcích dále přikládá elektronickou kopii zmocnění výrobcem se sídlem mimo členské
státy v českém nebo v anglickém jazyce.
2.3. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele,
přikládá tato osoba dále elektronickou kopii plné moci prokazující zmocnění k takovému
úkonu.
Příl.12
nadpis vypuštěn
1.
Formulář pro oznámení osoby nakládající s in vitro diagnostiky uvedené
v § 11 odst. 6 nařízení vlády č. 453/2004 Sb., ve znění nařízení vlády č. 67/2011
Sb.
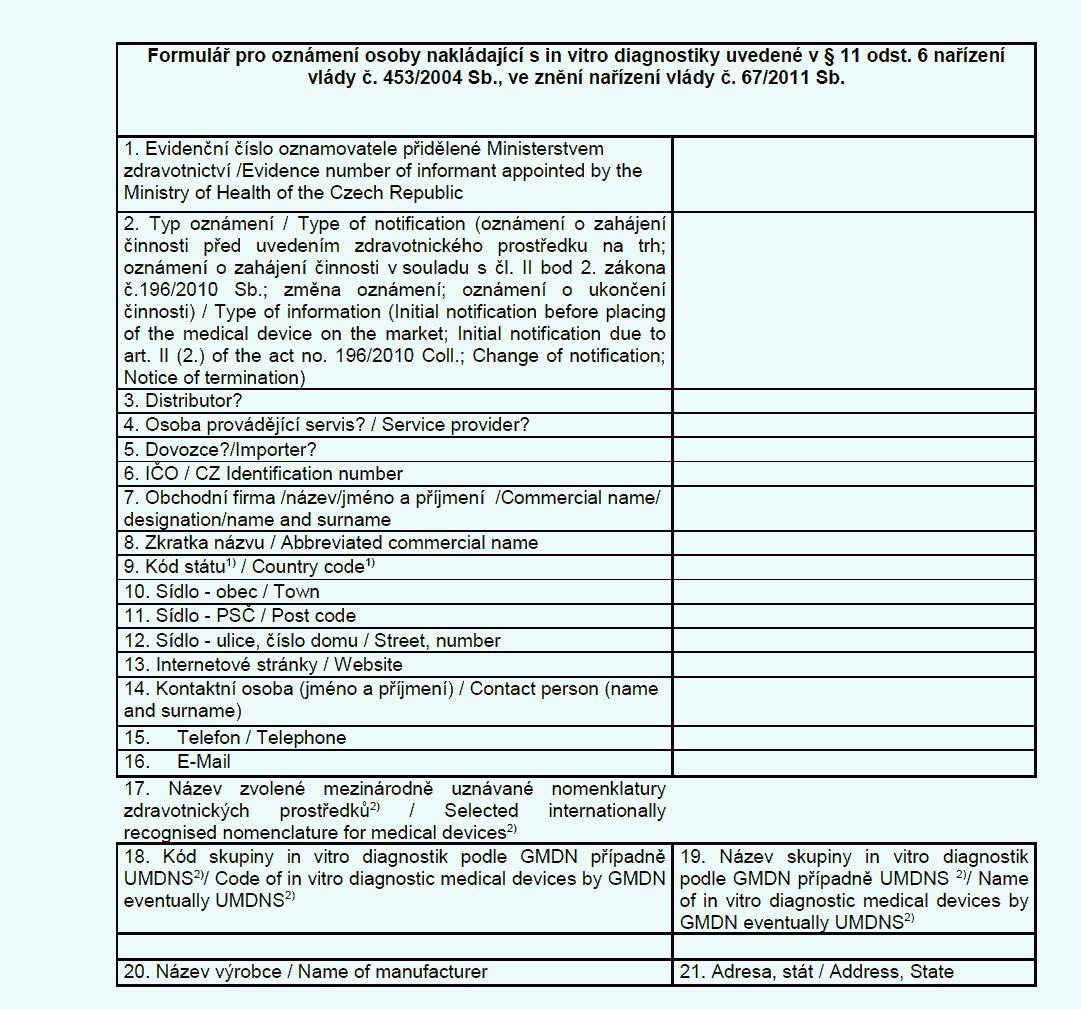
1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
1) Please
use the country codes according to EN ISO 3166- 1:2006, e.g.:
AT Rakousko / Austria,
BE Belgie / Belgium,
BG Bulharsko / Bulgaria,
CH Švýcarsko / Switzerland,
CY Kypr / Cyprus,
CZ Česká republika / Czech Republic,
DE Německo / Germany,
DK Dánsko / Denmark,
EE Estonsko / Estonia,
ES Španělsko / Spain,
FI Finsko / Finland,
FR Francie / France,
GB Spojené království / United Kingdom,
GR Řecko / Greece,
HU Maďarsko / Hungary,
IE Irsko / Ireland,
IS Island / Iceland,
IT Itálie / Italy,
LI Lichtenštejnsko / Lichtenstein,
LT Litva / Lithuania,
LU Lucembursko / Luxembourg,
LV Lotyšsko / Latvia,
MT Malta / Malta,
NL Nizozemsko / Netherlands,
NO Norsko / Norway,
PL Polsko / Poland,
PT Portugalsko / Portugal,
RO Rumunsko / Romania,
SE Švédsko / Sweden,
SI Slovinsko / Slovenia,
SK Slovensko / Slovakia.
2) Jestliže má výrobce, případně zplnomocněný zástupce, distribuovaného
in vitro diagnostika sídlo v jiném členském státě než České republice, který nevyžaduje
údaje podle Globální nomenklatury zdravotnických prostředků (GMDN), je možno uvést
údaje podle Univerzálního nomenklaturního systému zdravotnických (UMDNS).
2) If the
manufacturer or authorized representative of distributed in vitro diagnostic medical
device resides in other member state than Czech Republic, which does not require
information by Global Medical Device Nomenclature (GMDN), the distributor can fill
in information by Universal Medical Device Nomenclature System (UMDNS).
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1.
a způsob jeho vyplnění zveřejní Ministerstvo zdravotnictví na svých internetových
stránkách.
2.2. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele,
přikládá tato osoba dále elektronickou kopii plné moci prokazující zmocnění k takovému
úkonu.
Příl.13
nadpis vypuštěn
1.
Formulář pro oznámení in vitro diagnostika
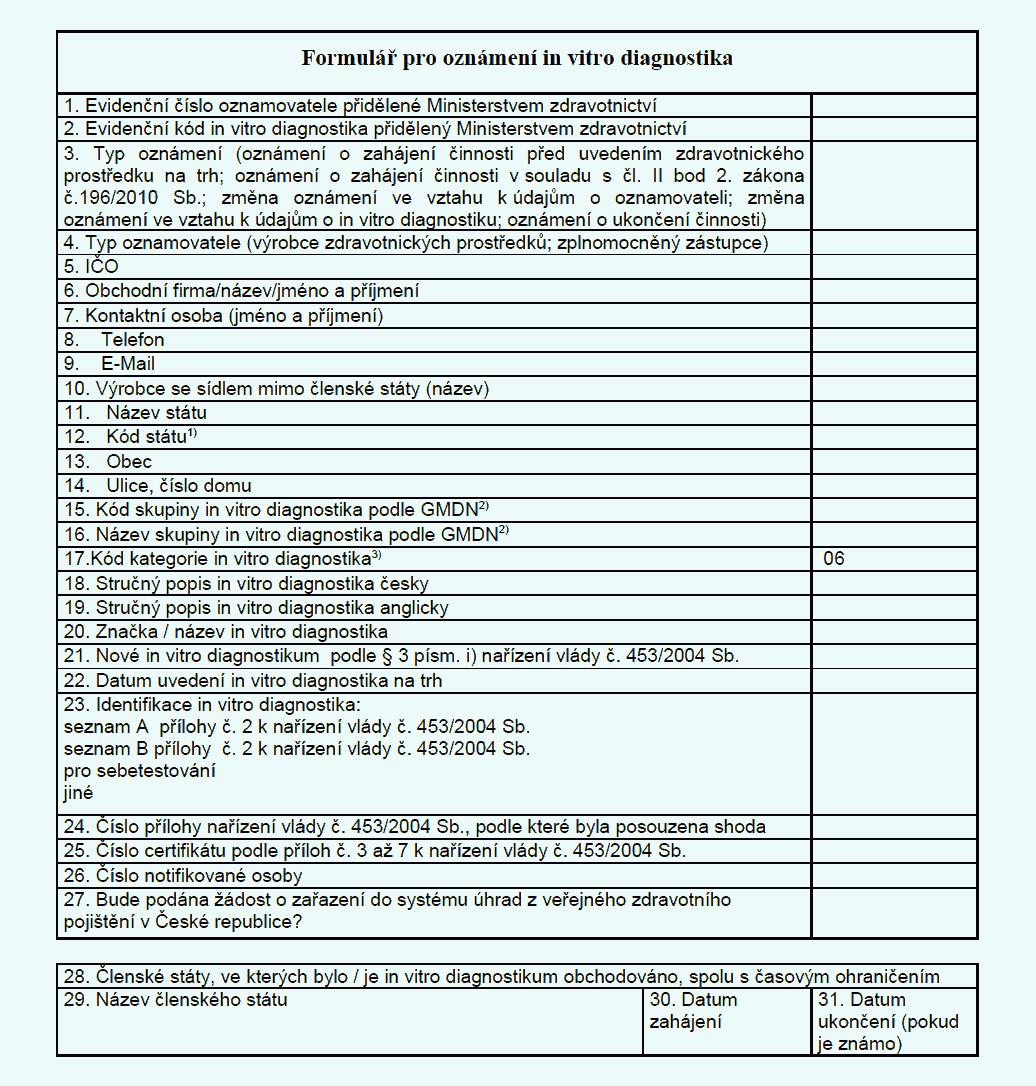
1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
AT Rakousko,
BE Belgie,
BG Bulharsko,
CH Švýcarsko,
CY Kypr,
CZ Česká republika,
DE Německo,
DK Dánsko,
EE Estonsko,
ES Španělsko,
FI Finsko,
FR Francie,
GB Spojené království,
GR Řecko,
HU Maďarsko,
IE Irsko,
IS Island,
IT Itálie,
LI Lichtenštejnsko,
LT Litva,
LU Lucembursko,
LV Lotyšsko,
MT Malta,
NL Nizozemsko,
NO Norsko,
PL Polsko,
PT Portugalsko,
RO Rumunsko,
SE Švédsko,
SI Slovinsko,
SK Slovensko.
2) Vyplňte správný kód a název skupiny in vitro diagnostika podle Globální
nomenklatury zdravotnických prostředků (GMDN).
3) ČSN EN ISO 15225:2010.
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1.
a způsob jeho vyplnění zveřejní Ministerstvo zdravotnictví na svých internetových
stránkách.
2.2. K formuláři se v elektronické podobě přikládají
2.2.1. informace vztahující se k
2.2.1.1. činidlům, výsledkům reakce
činidel, kalibračním a kontrolním materiálům se zřetelem na jejich obecnou technologickou
chrakteristiku nebo
2.2.1.2. analytům a všem jejich dalším podstatným změnám včetně
přerušení uvádění na trh,
2.2.1.3. dalším in vitro diagnostikům s odpovídajícími
údaji,
2.2.2. u in vitro diagnostik uvedených v příloze č. 2 k tomuto nařízení
a in vitro diagnostik pro sebetestování
2.2.2.1. údaje umožňující jejich identifikaci,
2.2.2.2. informace o jejich označování (například označení CE, štítky, symboly,
nápisy),
2.2.2.3. návody k jejich použití,
2.2.2.4. analytické a, přichází-li v
úvahu, i základní diagnostické údaje uvedené v bodě 3. části A. přílohy č. 1 k tomuto
nařízení,
2.2.2.5. výsledky hodnocení funkční způsobilosti podle přílohy č. 8 k
tomuto nařízení,
2.2.2.6. certifikáty a
2.2.2.7. podstatné změny vztahující se
k bodům 2.2.2.1. až 2.2.2.6. včetně přerušení uvádění na trh.
2.3. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele,
přikládá tato osoba dále elektronickou kopii plné moci prokazující zmocnění k takovému
úkonu.
1) Směrnice Evropského parlamentu a Rady 98/79/ES ze dne 27. října 1998 týkající
se diagnostických zdravotnických prostředků in vitro.
Rozhodnutí Komise 2002/364/ES ze 7. května 2002 o společných technických specifikacích
pro in vitro diagnostické zdravotnické prostředky.
Směrnice Komise 2011/100/EU ze dne 20. prosince 2011, kterou se mění směrnice
Evropského parlamentu a Rady 98/79/ES o diagnostických zdravotnických prostředcích
in vitro.
2) § 2 odst. 2 písm. c) zákona č. 123/2000 Sb., o zdravotnických prostředcích
a o změně některých souvisejících zákonů.
3) § 3 písm. e) zákona č. 123/2000 Sb., ve znění zákona č. 130/2003 Sb.
4) Nařízení vlády č. 336/2004 Sb., kterým se stanoví technické požadavky na
zdravotnické prostředky a kterým se mění nařízení vlády č. 251/2003 Sb., kterým
se mění některá nařízení vlády vydaná k provedení zákona č. 22/1997 Sb., o technických
požadavcích na výrobky a o změně a doplnění některých zákonů, ve znění pozdějších
předpisů.
5) § 4a zákona č. 22/1997 Sb., o technických požadavcích na výrobky a o změně
a doplnění některých zákonů, ve znění zákona č. 71/2000 Sb., zákona č. 102/2001
Sb., zákona č. 205/2002 Sb. a zákona č. 226/2003 Sb.
6) Rozhodnutí Evropské komise 2002/364/EC ze 7. května 2002 o společných technických
specifikacích pro in vitro diagnostické zdravotnické prostředky.
7) Úmluva na ochranu lidských práv a důstojnosti lidské bytosti v souvislosti
s aplikací biologie a medicíny: Úmluva o lidských právech a biomedicíně, vyhlášená
pod č. 96/2001 Sb. m. s.
8) Helsinská deklarace přijatá 18. Světovým zdravotnickým shromážděním v roce
1964 v Helsinkách ve Finsku, změněná a doplněná 29. Světovým lékařským shromážděním
v Tokiu v roce 1975, 35. Světovým lékařským shromážděním v Benátkách v roce 1983
a 41. Světovým lékařským shromážděním v Hong Kongu v roce 1989, v Somerset Westu
v Jihoafrické republice v roce 1996 a v Edinburgu v roce 2000.
9) Zákon č. 64/1986 Sb., o České obchodní inspekci, ve znění zákona č. 240/1992
Sb., zákona č. 22/1997 Sb., zákona č. 110/1997 Sb., zákona č. 189/1999 Sb., zákona
č. 71/2000 Sb., zákona č. 145/2000 Sb., zákona č. 102/2001 Sb., zákona č. 321/2001
Sb., zákona č. 205/2002 Sb., zákona č. 309/2002 Sb., zákona č. 226/2003 Sb. a zákona
č. 439/2003 Sb.
10) ČSN EN 980 "Značky pro označování zdravotnických prostředků".
11) § 3 písm. f) zákona č. 123/2000 Sb., ve znění zákona č. 130/2003 Sb.
12) Nařízení vlády č. 291/2000 Sb., kterým se stanoví grafická podoba označení
CE.
12a) Rozhodnutí Komise 2002/364/ES ze dne 7. května 2002 o společných technických
specifikacích pro diagnostické zdravotnické prostředky in vitro.
Rozhodnutí Komise 2009/108/ES ze dne 3. února 2009, kterým se mění rozhodnutí
2002/364/ES o společných technických specifikacích pro diagnostické zdravotnické
prostředky in vitro.
13) Například § 4a odst. 1 a 3 věta první zákona č. 22/1997 Sb., ve znění zákona
č. 205/2002 Sb. a zákona č. 277/2003 Sb.
14) § 11 odst. 7 zákona č. 22/1997 Sb., ve znění zákona č. 205/2002 Sb.
15) § 7 zákona č. 123/2000 Sb., ve znění zákona č.
16) § 3 písm. a) až c) zákona č. 64/1986 Sb., ve znění zákona č. 240/1992 Sb.
a zákona č. 145/2000 Sb.
17) § 7a zákona č. 64/1986 Sb., ve znění zákona č. 205/2002 Sb. a zákona č.
226/2003 Sb.
§ 19 odst. 1 zákona č. 22/1997 Sb., ve znění zákona č. 71/2000 Sb.,
zákona č. 205/2003 Sb. a zákona č. 226/2003 Sb.
18) Rozhodnutí Komise 2010/227/EU ze dne 19. 4. 2010 o Evropské databance zdravotnických
prostředků (Eudamed).
19) § 3 písm. g) a § 32 odst. 1 a odst. 3 písm. a) zákona 130/2001 Sb. č. 123/2000
Sb., ve znění zákona č. 130/2003 Sb.
Vyhláška č. 501/2000 Sb., kterou se stanoví
formy, způsoby ohlašování nežádoucích příhod zdravotnických prostředků, jejich
evidování, šetření a vyhodnocování, dokumentace a její uchovávání a následné sledování
s cílem předcházení vzniku nežádoucích příhod, zejména jejich opakování (vyhláška
o nežádoucích příhodách zdravotnických prostředků), ve znění vyhlášky č. 304/2003
Sb.
20) Příloha č. 1 k vyhlášce č. 304/2003 Sb., kterou se mění vyhláška č. 501/2000
Sb., v položce 2335 vedle kódu kategorie zdravotnického prostředku.
21) Vyhláška č. 501/2000 Sb., ve znění vyhlášky č. 304/2003 Sb.
22) § 41 zákona č. 123/2000 Sb., ve znění zákona č. 130/2003 Sb.
23) Zákon č. 258/2000 Sb., o ochraně veřejného zdraví a o změně některých souvisejících
zákonů, ve znění zákona č. 254/2001 Sb., zákona č. 274/2001 Sb., zákona č. 13/2002
Sb., zákona č. 76/2002 Sb., zákona č. 86/2002 Sb., zákona č. 120/2002 Sb., zákona
č. 309/2002 Sb., zákona č. 320/2002 Sb., zákona č. 274/2003 Sb., zákona č. 356/2003
Sb. a zákona č. 362/2003 Sb.
Zákon č. 123/2000 Sb., ve znění zákona č. 130/2003
Sb. a zákona č. 274/2003 Sb.
24) Zákon č. 102/2001 Sb., o obecné bezpečnosti výrobků a o změně některých
zákonů (zákon o obecné bezpečnosti výrobků), ve znění zákona č. 146/2002 Sb. a
zákona č. 277/2003 Sb.
Zákon č. 123/2000 Sb., ve znění pozdějších předpisů.
Zákon
č. 258/2000 Sb., ve znění pozdějších předpisů.
25) § 4 zákona č. 123/2000 Sb., ve znění zákonač. 130/2003 Sb.
26) Zákon č. 505/1990 Sb., o metrologii, ve znění zákona č. 4/1993 Sb., zákona
č. 20/1993 Sb., zákona č. 119/2000 Sb., zákona č. 13/2002 Sb., zákona č. 137/2002
Sb. a zákona č. 226/2003 Sb.
27) § 11 odst. 3 zákona č. 22/1997 Sb., ve znění zákona č. 71/2000 Sb.
28) § 11 odst. 2 písm. e) zákona č. 22/1997 Sb.