65/2011 Sb.
NAŘÍZENÍ VLÁDY
ze dne 23. února 2011,
kterým se mění nařízení vlády č. 336/2004 Sb., kterým se stanoví technické požadavky
na zdravotnické prostředky a kterým se mění nařízení vlády č. 251/2003 Sb.,
kterým se mění některá nařízení vlády vydaná k provedení zákona č. 22/1997 Sb.,
o technických požadavcích na výrobky a o změně a doplnění některých zákonů,
ve znění pozdějších předpisů, ve znění pozdějších předpisů
Vláda nařizuje podle § 22 zákona č. 22/1997 Sb.,
o technických požadavcích na výrobky a o změně a doplnění
některých zákonů, ve znění zákona č. 71/2000
Sb. a zákona č. 205/2002 Sb., (dále jen „zákon“) k provedení
§ 11 odst. 1 a 2, § 11a odst. 2, § 12 a 13 zákona
a k provedení zákona č. 123/2000 Sb., o zdravotnických
prostředcích a o změně některých souvisejících zákonů,
ve znění zákona č. 130/2003 Sb., zákona č. 274/2003
Sb., zákona č. 58/2005 Sb., zákona č. 227/2009 Sb. a zákona
č. 196/2010 Sb., (dále jen „zákon o zdravotnických
prostředcích“):
Čl. I
Nařízení vlády č. 336/2004 Sb., kterým se stanoví
technické požadavky na zdravotnické prostředky a kterým
se mění nařízení vlády č. 251/2003 Sb., kterým se
mění některá nařízení vlády vydaná k provedení zákona
č. 22/1997 Sb., o technických požadavcích na výrobky
a o změně a doplnění některých zákonů, ve znění pozdějších
předpisů, ve znění nařízení vlády č. 212/2007
Sb. a nařízení vlády č. 245/2009 Sb., se mění takto:
1. V § 1 úvodní části ustanovení, § 5 odst. 2 a v příloze
č. 12 bodech 1.2.7.1., 1.2.8.5. a 1.2.10. se slova
„Evropských společenství“ nahrazují slovy „Evropské
unie“.
2. V § 4 odst. 1 se věta druhá nahrazuje větou
„Existuje-li příslušné riziko, musí zdravotnické prostředky,
které jsou zároveň strojním zařízením8a), splňovat
rovněž základní požadavky na ochranu zdraví
a bezpečnost stanovené v příloze č. 1 k nařízení vlády
o technických požadavcích na strojní zařízení8a), pokud
jsou tyto základní požadavky na ochranu zdraví a bezpečnost
specifičtější než základní požadavky stanovené
v příloze č. 1 k tomuto nařízení.“.
3. V § 4 odst. 6 a v § 7 odst. 2 písm. e) se slova
„Komisi Evropských společenství“ nahrazují slovy
„Evropskou komisi“.
4. V § 6 odstavec 3 zní:
„(3) O opatřeních podle odstavců 1 a 2 informuje
Evropskou komisi a příslušné úřady členských států
Evropské unie nebo Evropského sdružení volného obchodu,
který je současně smluvní stranou Evropského
hospodářského prostoru, (dále jen „členský stát“) Ministerstvo
průmyslu a obchodu.“.
5. V § 9 odst. 5 se slova „(dále jen „ústav“)“ nahrazují
slovy „(dále jen „Ústav“)“.
6. V § 10 odst. 5, § 12 odst. 6 a 7, § 16 odst. 8,
příloze č. 1 bodě 7.4.2., příloze č. 2 bodě 8., příloze č. 4
bodě 9., příloze č. 5 bodě 7., příloze č. 10 bodě 2.3.5.
a v příloze č. 12 bodě 1.2.7.3. se slova „Evropských
společenství“ zrušují.
7. V § 11 odst. 5 se slova „v odstavcích 1 až 4“
nahrazují slovy „v odstavcích 2 a 4“.
8. § 13 a 14 včetně nadpisů znějí:
㤠13
Oznamovací povinnosti
(1) Výrobce, který v souladu s postupy podle § 4
odst. 3 uvádí zdravotnické prostředky na trh nebo do
provozu, zplnomocněný zástupce nebo osoba, která se
podílí na činnostech uvedených v § 11, oznamuje v elektronické
podobě ministerstvu
a) zahájení činnosti v souladu s přílohou č. 13 k tomuto
nařízení,
b) ukončení činnosti v souladu s přílohou č. 13 k tomuto
nařízení,
c) uvedení zdravotnického prostředku na trh v souladu
s přílohou č. 15 k tomuto nařízení, a to ještě
před uvedením zdravotnického prostředku na trh,
d) ukončení uvádění zdravotnického prostředku na
trh v souladu s přílohou č. 15 k tomuto nařízení a
e) změnu některého z údajů oznámených podle písmen
a) a c).
Údaje podle přílohy č. 15 k tomuto nařízení se oznamují
až poté, co oznamovatel obdrží od ministerstva
evidenční číslo na základě splnění oznamovací povinnosti
v souladu s přílohou č. 13 k tomuto nařízení.
Výrobce zakázkového zdravotnického prostředku
oznamuje v elektronické podobě ministerstvu pouze
údaje podle písmen a) a b) a změnu některého z údajů
oznámených podle písmene a).(2) Osoba, uvádějící zdravotnický prostředek na
trh nebo do provozu na území České republiky, je
povinna na žádost ministerstva poskytnout informace
umožňující identifikaci zdravotnického prostředku
a podklady, které sloužily k posouzení shody.
(3) Jestliže výrobce, který zamýšlí uvést pod svým
jménem na trh zdravotnický prostředek podle odstavce
1 nebo 2, nemá sídlo v členském státě, pověří
pro uvádění těchto zdravotnických prostředků na trh
zplnomocněného zástupce, který je jeho jediným
zplnomocněným zástupcem v rámci členských států.
(4) Ministerstvo na žádost sdělí členskému státu
a Evropské komisi informace vyplývající z odstavců 1
až 3 poskytnuté výrobcem nebo jeho zplnomocněným
zástupcem.
(5) Distributor, dovozce a osoba provádějící servis
zdravotnických prostředků oznamují v elektronické
podobě ministerstvu
a) zahájení činnosti v souladu s přílohou č. 14 k tomuto
nařízení,
b) ukončení činnosti v souladu s přílohou č. 14 k tomuto
nařízení a
c) změnu některého z údajů oznámených podle písmene
a).
§ 14
Soubor údajů
(1) Údaje
a) o zdravotnických prostředcích a osobách uvedených
v § 13,
b) o vydaných, změněných, pozastavených a zrušených
certifikátech, jakož i o zamítnutí žádostí o vydání
certifikátu podle postupů stanovených v přílohách
č. 2 až 7 k tomuto nařízení,
c) získané v souladu s postupem upravujícím oznamování
a evidenci nežádoucích příhod21) a
d) o klinických zkouškách
se zpracovávají v souladu s tímto nařízením v informačním
systému podle § 41 zákona o zdravotnických prostředcích
do doby, kdy osoba nakládající se zdravotnickými
prostředky oznámí ministerstvu ukončení činnosti
a po dobu dalších 20 let. Údaje z informačního
systému jsou přístupné ministerstvu, Úřadu pro
technickou normalizaci, metrologii a státní zkušebnictví
(dále jen „Úřad“), Ústavu, Státnímu úřadu pro jadernou
bezpečnost u zdrojů ionizujícího záření,
Ústavu zdravotnických informací a statistiky České
republiky a České obchodní inspekci. Údaje z informačního
systému o nežádoucích příhodách22) a klinických
zkouškách zdravotnických prostředků jsou přístupné
pouze ministerstvu, Ústavu a Ústavu zdravotnických
informací a statistiky České republiky
a u zdrojů ionizujícího záření Státnímu úřadu pro
jadernou bezpečnost.(2) Údaje uvedené v odstavci 1 písm. a) a d) se
poskytují v elektronické podobě podle příloh č. 13
až 16 k tomuto nařízení.“.
9. V § 15 odstavec 1 zní:
„(1) U zdravotnických prostředků určených pro
klinické zkoušky výrobce, zplnomocněný zástupce
nebo zadavatel postupuje podle přílohy č. 8 k tomuto
nařízení a oznamuje Ústavu záměr provést klinické
zkoušky formou stanovenou v příloze č. 16 k tomuto
nařízení a současně předkládá prohlášení včetně dokumentace
uvedené v bodě 2.2. přílohy č. 8 k tomuto nařízení.“.
10. V § 15 odst. 2 se slovo „ministerstvo“ nahrazuje
slovem „Ústav“.
11. V § 15 odst. 2 a 5 se slovo „ministerstva“ nahrazuje
slovem „Ústavu“.
12. V § 15 odstavec 6 zní:
„(6) Výrobce, zplnomocněný zástupce nebo zadavatel
oznámí Ústavu ukončení klinické zkoušky, včetně
jeho odůvodnění v případě předčasného ukončení. Je-li
klinická zkouška předčasně ukončena z bezpečnostních
důvodů, předá Ústav toto oznámení všem členským
státům a Evropské komisi. Výrobce nebo zplnomocněný
zástupce uchovává zprávu uvedenou v § 11
písm. c) zákona o zdravotnických prostředcích k dispozici
příslušným orgánům.“.
13. V § 16 odst. 5 se slova „a ministerstvo“ zrušují.
14. V § 16 odst. 6 se za slova „jiných“ vkládají
slova „členských“ a slova „Komise Evropských společenství“
se nahrazují slovy „Evropské komise“.
15. V příloze č. 1 bod 7.1.2. zní:
„7.1.2. vzájemnou kompatibilitu mezi použitými materiály a biologickými tkáněmi,
buňkami a tělesnými
tekutinami se zřetelem na určený účel použití,“.
16. V příloze č. 1 bodech 7.4.2. a 7.4.5. se slovo
„ústav“ nahrazuje slovem „Ústav“.
17. V příloze č. 1 bod 7.4.4. zní:
„7.4.4. Jsou-li prováděny změny na doplňující látce začleněné do zdravotnického prostředku,
zejména pokud jde o její výrobní postup, musí být o změnách informována notifikovaná
osoba, která požádá o odborné stanovisko odpovídající příslušný úřad pro léčivé přípravky
(tj. úřad, který vydal původní odborné stanovisko), aby bylo potvrzeno, že jakost
a bezpečnost doplňující látky je zachována. Je-li o odborné stanovisko požádán Ústav,
přihlíží k údajům o užitečnosti začlenění doplňující látky do zdravotnického prostředku,
které uvedla notifikovaná osoba za účelem zajištění, že změny nemají žádný negativní
dopad na stanovené riziko při začlenění této doplňující látky do zdravotnického prostředku.“.
18. V příloze č. 1 se za bod 12.1. vkládá nový
bod 12.1a., který zní:
„12.1a. U zdravotnických prostředků, které obsahují programové vybavení nebo které
jsou samy o sobě
zdravotnickým programovým vybavením, musí být programové vybavení validováno podle
nejnovějších
poznatků s přihlédnutím k zásadám vývoje životního cyklu, řízení rizika, validace
a ověřování.“.
19. V příloze č. 1 bodě 12.9. se slova „pokud je to
vhodné,“ nahrazují slovy „podle potřeby“.
20. V příloze č. 1 bod 13.3.1. zní:
„13.3.1. jméno, popřípadě jména a příjmení výrobce, adresu jeho místa podnikání,
jestliže výrobcem
je fyzická osoba; název nebo obchodní firmu, adresu sídla, jestliže výrobcem je
právnická osoba; u zdravotnických prostředků dovážených do Evropské unie s předpokladem
jejich distribuce v rámci Evropské unie musí etiketa nebo štítek, vnější obal nebo
návod k použití navíc obsahovat jméno, popřípadě jména a příjmení nebo název nebo
obchodní firmu a adresu místa podnikání nebo sídla zplnomocněného zástupce, jestliže
výrobce nemá sídlo v členském státě,“.
21. V příloze č. 1 bod 13.3.6. zní:
„13.3.6. případně označení, že se jedná o zdravotnický prostředek pro jednorázové
použití.
Informace výrobce o tom, že se jedná o zdravotnický prostředek pro jednorázové použití,
musí být v rámci Evropské unie jednotná,“.
22. V příloze č. 1 bodě 13.6.8. se slova „pro 1 použití“
nahrazují slovy „pro jednorázové použití“.
23. V příloze č. 1 bodě 13.6.17. se za slovo „vydání“
vkládá slovo „nebo“.
24. V příloze č. 2 bodě 3.1.7., příloze č. 4 bodě 3.,
příloze č. 5 bodě 3.1.1.8., příloze č. 7 bodě 4. a v příloze
č. 8 bodě 5. se slovo „ústavu“ nahrazuje slovem
„Ústavu“.
25. V příloze č. 2 bod 4.3.2. zní:
„4.3.2. V případě zdravotnických prostředků uvedených v bodě 7.4.1. přílohy č. 1
k tomuto
nařízení požádá notifikovaná osoba, v souladu s bodem 7.4.2. přílohy č. 1 k tomuto
nařízení, před přijetím rozhodnutí o odborné stanovisko jeden z příslušných úřadů
členských
států, v České republice Ústav, nebo EMEA. Je-li o odborné stanovisko požádán
Ústav, vypracuje odborné stanovisko do 210 dnů ode dne obdržení úplné dokumentace.
Odborné stanovisko příslušného úřadu členského státu nebo EMEA musí být zahrnuto
do dokumentace týkající se zdravotnického prostředku. Notifikovaná osoba při přijímání
rozhodnutí věnuje tomuto odbornému stanovisku náležitou pozornost a své konečné
rozhodnutí sdělí instituci, která toto odborné stanovisko vydala.“.
26. V příloze č. 2 se bod 6.2. zrušuje.
27. V příloze č. 3 bodě 3.6. a v příloze č. 8 bodě
3.2.5. se za slovo „účelu“ vkládá slovo „použití“.
28. V příloze č. 3 bod 5.2. zní:
„5.2. V případě zdravotnických prostředků uvedených v bodě 7.4.1. přílohy č. 1 k
tomuto nařízení
požádá notifikovaná osoba, v souladu s bodem 7.4.2. přílohy č. 1 k tomuto nařízení,
před přijetím
rozhodnutí o odborné stanovisko jeden z příslušných úřadů členských států, v České
republice
Ústav, nebo EMEA. Je-li o odborné stanovisko požádán Ústav, vypracuje odborné stanovisko
do
210 dnů ode dne obdržení úplné dokumentace. Odborné stanovisko příslušného úřadu
členského
státu nebo EMEA musí být zahrnuto do dokumentace týkající se zdravotnického prostředku.
Notifikovaná osoba při přijímání rozhodnutí věnuje tomuto odbornému stanovisku náležitou
pozornost a své konečné rozhodnutí sdělí instituci, která toto odborné stanovisko
vydala.“.
29. V příloze č. 8 bod 2.2.9. zní:
„2.2.9. název právnické osoby nebo jméno, popřípadě jména a příjmení fyzické osoby,
která zadává
provedení klinické zkoušky, a jméno, popřípadě jména a příjmení lékaře nebo jiné
pověřené osoby, která prakticky provádí klinickou zkoušku; na tyto osoby se vztahují
povinnosti a odpovědnost uvedené v § 8 až 14 zákona o zdravotnických prostředcích,“.
30. V příloze č. 9 bod 1.4. zní:
„1.4. „aktivní zdravotnický prostředek“ znamená zdravotnický prostředek, jehož činnost
závisí na
zdroji elektrické nebo jiné energie, která není přímo vytvářena lidským tělem nebo
gravitací a který
působí prostřednictvím přeměny této energie; zdravotnické prostředky určené k přenosu
energie
nebo látek mezi aktivním zdravotnickým prostředkem a pacientem bez jakékoliv významné
změny
se za aktivní zdravotnický prostředek nepovažují; samostatné programové vybavení
se považuje za
aktivní zdravotnický prostředek,“.
31. V příloze č. 10 bodě 1.3. se slovo „řádné“ nahrazuje
slovem „plné“.
32. V příloze č. 10 bodě 1.6. se za slovo „považují“
vkládají slova „v souladu s ustanovením § 49 zákona
o zdravotnických prostředcích“.
33. V příloze č. 10 bodě 2.3.5. se slova „nepříznivé
události“ nahrazují slovy „nežádoucí příhody“.
34. V příloze č. 10 bodě 3. se slova „§ 12 odst. 1
písm. e) a § 12 odst. 2 písm. c)“ nahrazují slovy „§ 11
písm. c)“.
35. Přílohy č. 13 až 16 znějí:
„Příloha č. 13 k nařízení vlády č. 336/2004 Sb.
Formulář pro oznámení osoby nakládající se zdravotnickými prostředky
uvedené v § 13 odst. 1 nařízení vlády č. 336/2004 Sb.,
ve znění nařízení vlády č. 65/2011 Sb.

1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
AT Rakousko,
BE Belgie,
BG Bulharsko,
CH Švýcarsko,
CY Kypr,
CZ Česká republika,
DE Německo,
DK Dánsko,
EE Estonsko,
ES Španělsko,
FI Finsko,
FR Francie,
GB Spojené království,
GR Řecko,
HU Maďarsko,
IE Irsko,
IS Island,
IT Itálie,
LI Lichtenštejnsko,
LT Litva,
LU Lucembursko,
LV Lotyšsko,
MT Malta,
NL Nizozemsko,
NO Norsko,
PL Polsko,
PT Portugalsko,
RO Rumunsko,
SE Švédsko,
SI Slovinsko,
SK Slovensko.
2) Vyplňte správný kód a název skupiny zdravotnického prostředku podle Globální nomenklatury
zdravotnických prostředků (GMDN).
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1. a způsob jeho
vyplnění zveřejní Ministerstvo zdravotnictví na svých internetových stránkách.
2.2. Zplnomocněný zástupce podle § 3 písm. l) zákona o zdravotnických prostředcích
dále přikládá elektronickou kopii zmocnění výrobcem se sídlem mimo členské státy
v českém nebo v anglickém jazyce.
2.3. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele, přikládá
tato osoba dále elektronickou kopii plné moci prokazující zmocnění k takovému úkonu.
Příloha č. 14 k nařízení vlády č. 336/2004 Sb.
Formulář pro oznámení osoby nakládající se zdravotnickými prostředky
uvedené v § 13 odst. 5 nařízení vlády č. 336/2004 Sb.,
ve znění nařízení vlády č. 65/2011 Sb.
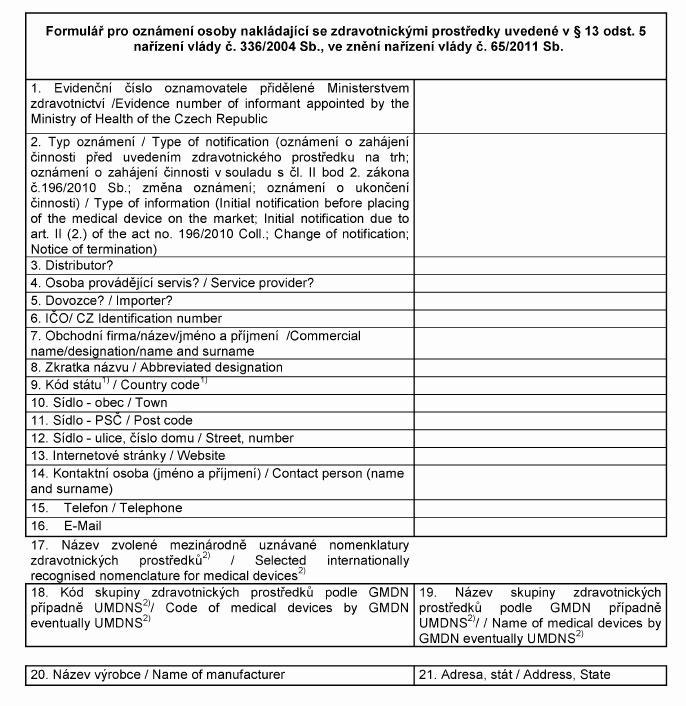
1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
1) Please use the country codes according to EN ISO 3166-1:2006, e.g.:
AT Rakousko / Austria,
BE Belgie / Belgium,
BG Bulharsko / Bulgaria,
CH Švýcarsko / Switzerland,
CY Kypr / Cyprus,
CZ Česká republika / Czech Republic,
DE Německo / Germany,
DK Dánsko / Denmark,
EE Estonsko / Estonia,
ES Španělsko / Spain,
FI Finsko / Finland,
FR Francie / France,
GB Spojené království / United Kingdom,
GR Řecko / Greece,
HU Maďarsko / Hungary,
IE Irsko / Ireland,
IS Island / Iceland,
IT Itálie / Italy,
LI Lichtenštejnsko / Lichtenstein,
LT Litva / Lithuania,
LU Lucembursko / Luxembourg,
LV Lotyšsko / Latvia,
MT Malta / Malta,
NL Nizozemsko / Netherlands,
NO Norsko / Norway,
PL Polsko / Poland,
PT Portugalsko / Portugal,
RO Rumunsko / Romania,
SE Švédsko / Sweden,
SI Slovinsko / Slovenia,
SK Slovensko / Slovakia.
2) Jestliže má výrobce, případně zplnomocněný zástupce, distribuovaného zdravotnického
prostředku sídlo v jiném členském státě než České republice, který nevyžaduje údaje
podle Globální
nomenklatury zdravotnických prostředků (GMDN), je možno uvést údaje podle Univerzálního
nomenklaturního systému zdravotnických prostředků (UMDNS).
2) If the manufacturer or authorized representative of distributed medical device
resides in
other member state than Czech Republic, which does not require information by Global
Medical Device
Nomenclature (GMDN), the distributor can fill in information by Universal Medical
Device
Nomenclature System (UMDNS).
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1. a způsob jeho
vyplnění
zveřejní Ministerstvo zdravotnictví na svých internetových stránkách.
2.2. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele, přikládá
tato osoba
dále elektronickou kopii plné moci prokazující zmocnění k takovému úkonu.
Příloha č. 15 k nařízení vlády č. 336/2004 Sb.
Formulář pro oznámení zdravotnického prostředku
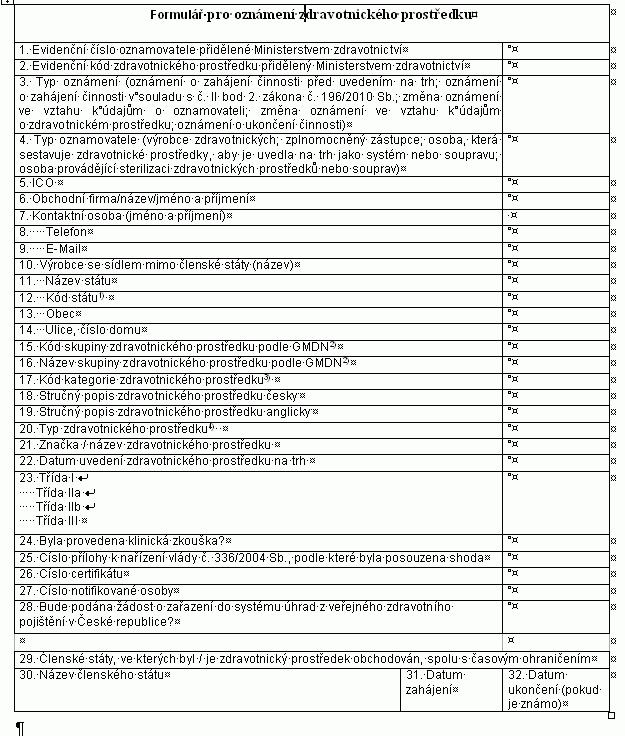
1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
AT Rakousko,
BE Belgie,
BG Bulharsko,
CH Švýcarsko,
CY Kypr,
CZ Česká republika,
DE Německo,
DK Dánsko,
EE Estonsko,
ES Španělsko,
FI Finsko,
FR Francie,
GB Spojené království,
GR Řecko,
HU Maďarsko,
IE Irsko,
IS Island,
IT Itálie,
LI Lichtenštejnsko,
LT Litva,
LU Lucembursko,
LV Lotyšsko,
MT Malta,
NL Nizozemsko,
NO Norsko,
PL Polsko,
PT Portugalsko,
RO Rumunsko,
SE Švédsko,
SI Slovinsko,
SK Slovensko.
2) Vyplňte správný kód a název skupiny zdravotnického prostředku podle Globální nomenklatury
zdravotnických prostředků (GMDN).
3) ČSN EN ISO 15225:2010. Zdravotnický prostředek se označuje kódem první vhodné
kategorie, do které spadá.
4) Určitý výrobní model nebo varianta modelu podle specifikace uvedené v certifikátu.
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1. a způsob jeho
vyplnění
zveřejní Ministerstvo zdravotnictví na svých internetových stránkách.
2.2. K formuláři se přikládá elektronická kopie závěrečné zprávy z klinického hodnocení,
certifikátu (byl-li vydán) a prohlášení o shodě v českém nebo anglickém jazyce.
2.3. K formuláři se přikládá elektronická kopie návodu k použití v českém jazyce.
2.4. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele, přikládá
tato osoba
dále elektronickou kopii plné moci prokazující zmocnění k takovému úkonu.
Příloha č. 16 k nařízení vlády č. 336/2004 Sb.
Formulář o záměru provést klinické zkoušky
Clinical Investigation Notification Form
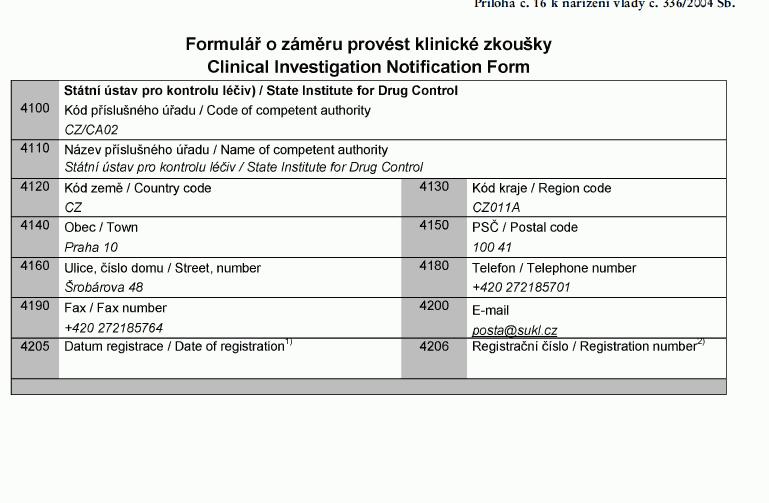
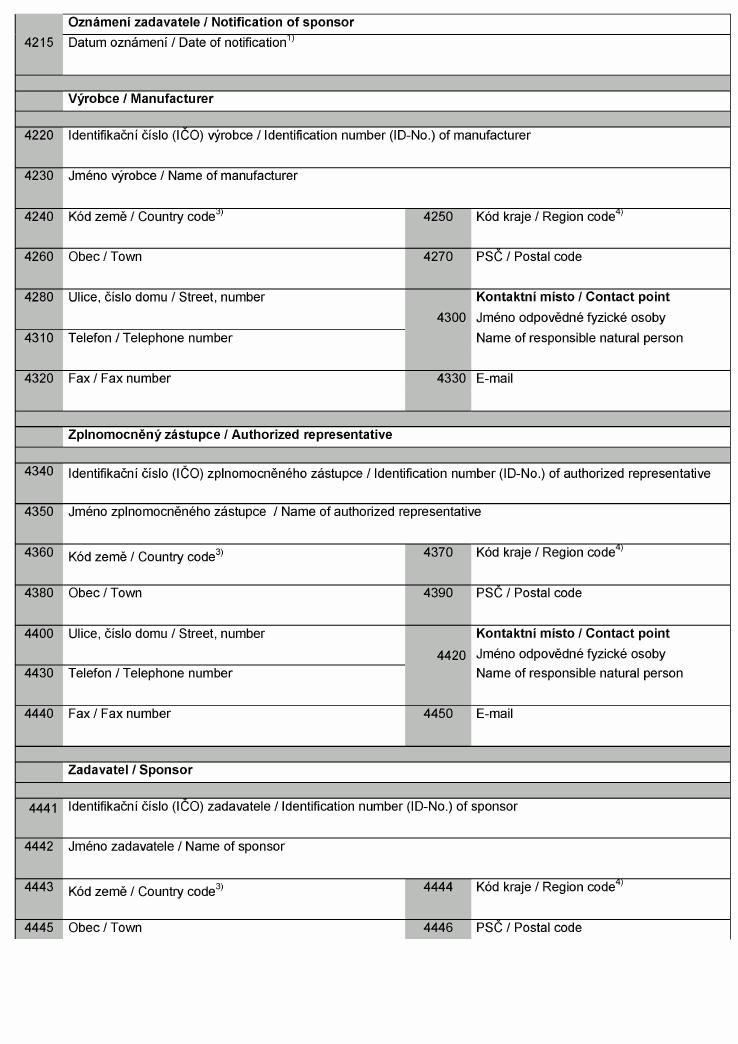
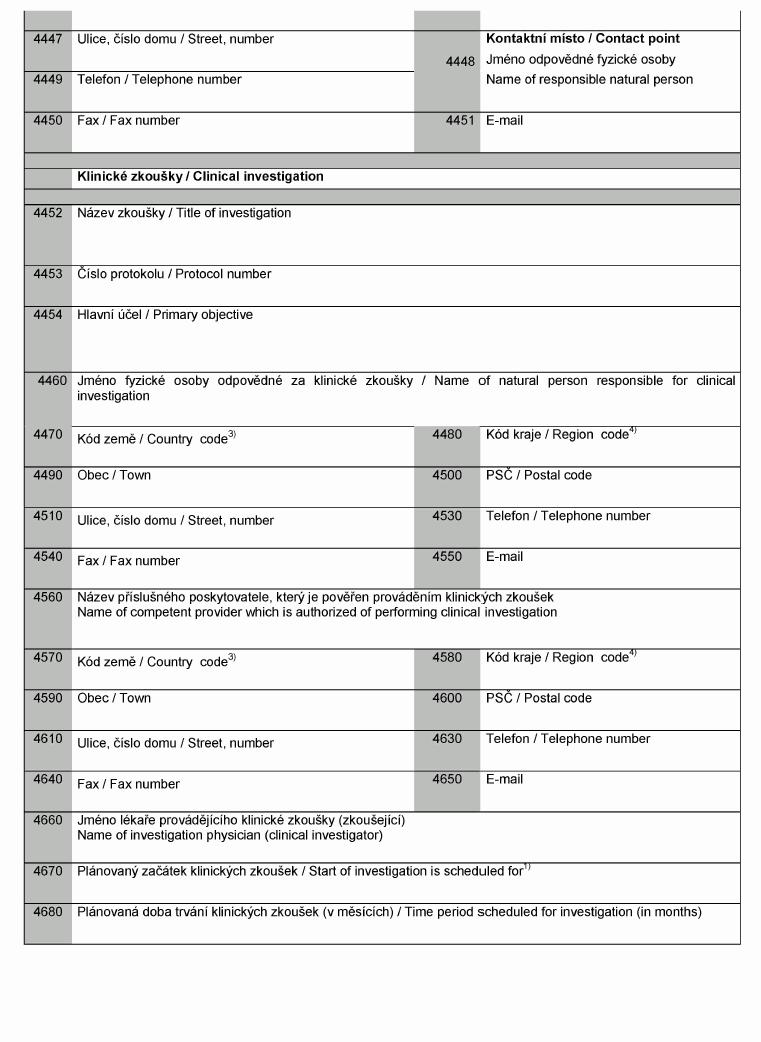

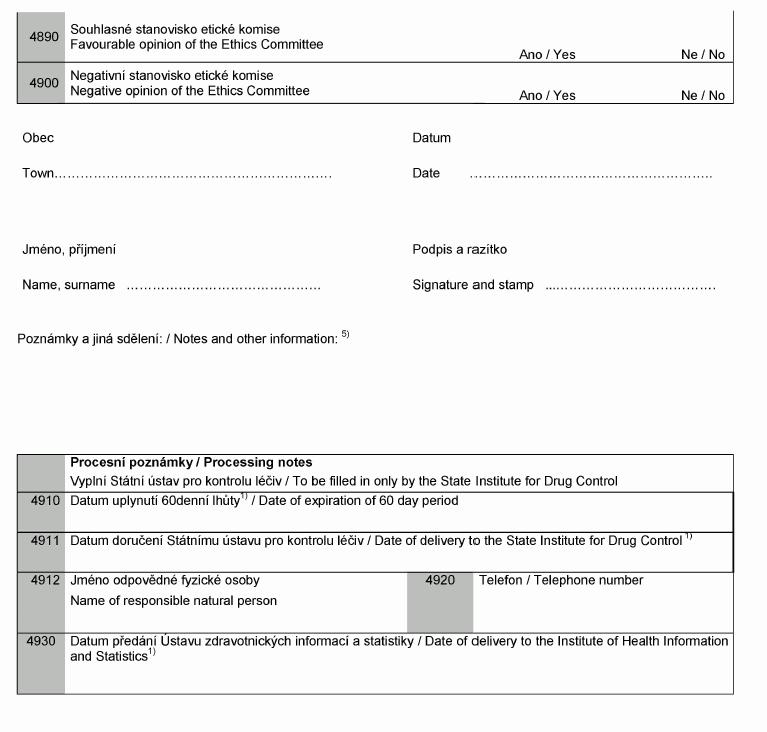
1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
AT Rakousko,
BE Belgie,
BG Bulharsko,
CH Švýcarsko,
CY Kypr,
CZ Česká republika,
DE Německo,
DK Dánsko,
EE Estonsko,
ES Španělsko,
FI Finsko,
FR Francie,
GB Spojené království,
GR Řecko,
HU Maďarsko,
IE Irsko,
IS Island,
IT Itálie,
LI Lichtenštejnsko,
LT Litva,
LU Lucembursko,
LV Lotyšsko,
MT Malta,
NL Nizozemsko,
NO Norsko,
PL Polsko,
PT Portugalsko,
RO Rumunsko,
SE Švédsko,
SI Slovinsko,
SK Slovensko.
2) Vyplňte správný kód a název skupiny zdravotnického prostředku podle Globální nomenklatury
zdravotnických prostředků (GMDN).
3) ČSN EN ISO 15225:2010. Zdravotnický prostředek se označuje kódem první vhodné
kategorie, do které spadá.
4) Určitý výrobní model nebo varianta modelu podle specifikace uvedené v certifikátu.
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1. a způsob jeho
vyplnění zveřejní Ministerstvo zdravotnictví na svých internetových stránkách.
2.2. K formuláři se přikládá elektronická kopie závěrečné zprávy z klinického hodnocení,
certifikátu (byl-li vydán) a prohlášení o shodě v českém nebo anglickém jazyce.
2.3. K formuláři se přikládá elektronická kopie návodu k použití v českém jazyce.
2.4. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele, přikládá
tato osoba
dále elektronickou kopii plné moci prokazující zmocnění k takovému úkonu.
Čl. II
Účinnost
Toto nařízení nabývá účinnosti dnem 1. dubna
2011.
Předseda vlády:
RNDr. Nečas v. r.
Ministr zdravotnictví:
doc. MUDr. Heger, CSc., v. r.